Technologie pro sekvenování DNA byla vyvinuta již v roce 1977 díky Fredericku Sangerovi. Trvalo to trochu déle, než bylo možné sekvenovat kompletní genom. Je to proto, že jsme potřebovali vhodný matematický model a obrovskou výpočetní sílu, abychom shromáždili miliony nebo miliardy malých čtení do většího kompletního genomu. Dnešní výpočetní výkon a software jsou hlavním rozdílem mezi tím, co trvalo roky práce na počátku roku 2000, a tím, co dnes trvá jen několik hodin. Algoritmus, který jste se rozhodli udělat, je „svatý grál“ montážní technologie. Tyto algoritmy obsahují jednu z nejznámějších proměnných známých v matematických modelech, k-mer.
původ k-mer a matematický model, který jej obklopuje, pochází od švýcarského matematika Leonharda Eulera z roku 1735, který je známý jako otec matematické funkce. Nizozemský matematik Nicolaas de Bruijn přizpůsobil Eulerovy myšlenky, aby našel cyklickou posloupnost písmen převzatých z dané abecedy, pro kterou se každé možné slovo určité délky objeví jako řetězec po sobě jdoucích znaků v cyklické sekvenci přesně jednou.
de Bruijnův algoritmus byl upraven molekulárními biology, kteří o mnoho let později čelili ekvivalentnímu problému: jak sestavit sekvence DNA. Vědci z celého světa tak nyní používají de Bruijnův graf a proměnnou k.
aplikace k-mers na sestavení sekvencí DNA
v několika slovech zahrnuje de novo sestavení genomu připojení po sobě jdoucích malých čtení DNA a ukončení větších sekvencí. Pro generování de Bruijnova grafu (viz obrázek níže) musí nukleotidy na okraji každého čtení překrývat okraj druhého (a tak dále). Konečným cílem je vytvořit po sobě jdoucí vrchol, který (potenciálně) povede k velkým fragmentům DNA.
musíte fragmentovat vaše čtení do k-mers, což je specifický počet nukleotidů, které se překrývají. K-mer umožňuje generovat jedinečnou sekvenci z mnoha malých. Každá jedinečná sekvence k-mer je identifikována a další kopie jsou odstraněny. Tento aspekt k-mers vám umožňuje překonat jednu z nevýhod sekvenování nové generace-získání čtení, které představují genomické oblasti s různými frekvencemi (tj. Použití k-mers eliminuje sekvence opakované více než jednou kvůli nerovnému pokrytí sekvencí. Mějte však na paměti, že nízká velikost k-mer zvýší šance na překrývání nukleotidů, zatímco jejich větší hodnota je sníží.
dnešní technologie montáže de novo je efektivnější, když používáte knihovny velkých čtení (tj. 1.000–10.000 bps) v kombinaci s menšími (100-200 bps). Softwarové programy mohou používat hodnotu k a k-mers pro sestavení krátkých čtení. Ty pak mohou být začleněny a ověřeny většími, aby skončily v přesnějších souvislostech.
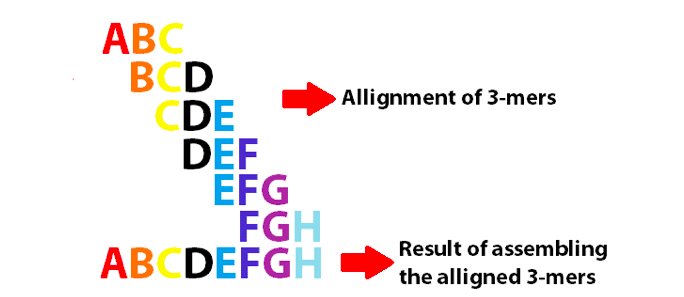
příklad de Bruijnova grafu pomocí 3-mers k sestavení 8 prvních písmen anglické abecedy. Všimněte si, že tyto 3-Mery se překrývají jako k-1.
čím více víte, tím více můžete dosáhnout v sestavě DNA
existují konkrétní tipy, které je třeba zvážit před použitím grafů De Bruijn v metodě montáže a výběrem nejvhodnější velikosti k-mer. Jejich využitím můžete dosáhnout lepších výsledků.
- nejprve a možná nejdůležitější je použití mnoha různých k-mers ve vaší sestavě. Pak byste měli vyhodnotit své výsledky a vybrat ten nejlepší (y). Nikdy nezapomeňte, že téměř nikdy neexistuje jedna a jediná správná sestava.
- před použitím k-mer byste měli pečlivě zpracovat čtení chyb. Pokud chyby pečlivě neodstraníte, výsledky mohou vytvořit nežádoucí vyboulení, což komplikuje montáž. Zvyšte prahovou hodnotu pro chybovost, kterou používáte při ořezávání sekvencí. Můžete ztratit některé sekvence, ale ti, kteří zůstanou, budou nejlepší.
- s opakováním DNA byste měli zacházet opatrně. Například sekvenování Illumina generuje velmi velké množství dat. Nejprve se pokuste sestavit malý zlomek čtení a poté je všechny použít k odhalení rozdílů. Opakovatelné krátké čtení může negativně ovlivnit proces montáže.
- znát Vaše data. Pokud neznáte velikost očekávaného genomu, množství sekvenačního pokrytí a počet čtení, pak jste náchylnější zvolit nejlepší hodnotu k pro sestavení genomu. Můžete navštívit poradce k-mer, jako velvet advisor z Monash university, abyste získali radu, která hodnota se zdá být vhodnější.
použití k-mers různých délek a zarovnání spojů také pomáhá vědcům zjistit míru mutace a rozšířit její použití. Manipulace s de Bruijnovými grafy směrem k přínosu montáže samozřejmě není všelékem. Existuje mnoho věcí, které je třeba zvážit, než zjednodušující funkce pro sestavení genomu živého organismu. Toto je jen úvod do historie a jak ji biologové mohou efektivněji využívat.
- Compeau PE, Pevzner PA, Tesler G. (2011). Jak aplikovat de Bruijn grafy na sestavení genomu.Přírodní Biotechnologie. 29(11):987–91.
- Aggarwala V, Voight BF. (2016). Rozšířený sekvenční kontextový model široce vysvětluje variabilitu úrovní polymorfismu napříč lidským genomem. Přírodní Genetika. 48(4): 349–55.

pomohlo vám to? Pak prosím sdílejte s vaší sítí.