Úvod
Gen adheze molekul buněk L1 (L1CAM) je molekula adheze neuronálních buněk patřící do nadčeledi imunoglobulinů; má klíčové funkce ve vývoji nervového systému (Itoh a Fushiki, 2015). Mutace v L1CAM souvisejí s X-vázanými neurologickými syndromy, které jsou shrnuty jako nemoci L1. Jsou klasifikovány takto: X-vázaný hydrocefalus (XLH) způsobený stenózou akvaduktu Sylvius (HSAS), MASA syndrom (mentální postižení, afázie, míchání chůze, aduktované palce), spastická paraparéza typu 1 (SP1) a X-vázaná ageneze corpus callosum (ACC) (Weller a Gartner, 2001; Itoh a Fushiki, 2015).
v hgmd® Professional 2019.2 (https://portal.biobase-international.com/hgmd/pro/all.php) bylo hlášeno asi 282 mutací způsobujících onemocnění (DMs) v genu L1CAM. Změny v genu L1CAM jsou různé; analýzy mutačních dat od 282 pacientů odhalují 51% chybných a nesmyslných mutací, 25% delecí,5% inzercí a 19% změn v místě spojení, ale tiché mutace v L1CAM s patogenním potenciálem byly vzácné a tiché mutace byly často ignorovány zejména při detekci sekvenování celých exomů (WES).
v této studii jsme pomocí WES prověřili fetální DNA čínské těhotné ženy, která hlásila pět nepřetržitých těhotenství s fetálním hydrocefalem; našli jsme pouze novou tichou mutaci c. 453G > T (s. Gly151 = ) v genu L1CAM. Zajímavé je, že další analýzou jsme naznačili, že tichá mutace vytvořila potenciální sekvenci konsensu 5 ‚ splice site, což by mělo za následek in-frame deleci 72 bp z exonu 5 a 24 aminokyseliny proteinu L1CAM.
prezentace případu
28letá zdravá žena byla předána na naši kliniku po čtyřech dobrovolných ukončení těhotenství v důsledku fetálního hydrocefalu v jiných nemocnicích. Všechny plody byly mužské. Když dorazila do naší nemocnice (ženská nemocnice, lékařská fakulta, Zhejiang University, Zhejiang, Čína), byla již na svém pátém těhotenství ve 24 týdnech těhotenství, s fetálním hydrocefalem obrazovými vyšetřeními. K prozkoumání genetické příčiny byl proveden odběr krve plodu ve 26 týdnech gestačního věku. Byly provedeny konvenční cytogenetické studie pro fetální i rodičovské vzorky a vzorek plodu byl dále analyzován pomocí pole s jedním nukleotidovým polymorfismem (SNP) a WES.
tato studie byla provedena v souladu s doporučeními Etické komise ženské nemocnice, lékařské fakulty Zhejiang University a informovaný souhlas byl získán od všech účastníků této studie v souladu s Helsinskou deklarací. Protokol studie byl schválen revizní komisí ženské nemocnice, lékařské fakulty, Zhejiang University v Číně.
materiály a metody
karyotyp a SNP pole
karyotypy fetální pupečníkové krve a periferní pupečníkové krve byly stanoveny konvenčním karyotypizací nejméně 30 krevních lymfocytů, které byly zastaveny při metafáze kolchiciny. G-bandážní karyotypy kultivovaných buněk byly provedeny na úrovni 320-400 pásem s rozlišením přibližně 10 Mb. SNP array bylo provedeno CytoScan™ HD array (Affymetrix, USA) podle pokynů výrobce, s přibližně 2,600,000 markery včetně 750,000 SNP sond a 1,900,000 nepolymorfních sond pro komplexní pokrytí celého genomu. Data byla analyzována pomocí softwaru chromozom Analysis Suite (ChAS) (Affymetrix, Santa Clara, CA) založeného na sestavě GRCh37/hg19. Práh vykazování výsledku počtu kopií byl stanoven na 500 kb s počtem markerů ≥50 pro zisky a na 200 kb s počtem markerů ≥50 pro ztráty.
sekvenování celého Exomu
hlavní část WES poskytl Pekingský genomický Institut. Genomická DNA byla extrahována krevní soupravou DNeasy (Qiagen, CA) a poté byla fragmentována Covaris LE220 (Massachusetts, USA), aby se vytvořila párová knihovna (200-250 bp). Všechny amplifikované knihovny byly provedeny na platformě BGISEQ-500, jednovláknová DNA byla smíchána s Mgieasy™ DNA Library Prep Kit V1 (BGI, Shenzhen, Čína) a poté sekvenována pomocí chemie 100SR s vysoce výkonnou sekvenční sadou BGISEQ-500RS (BGI, Shenzhen, Čína).
čisté čtení (s délkou 90 bp) odvozené z cíleného sekvenování a filtrování byly poté zarovnány s referencí lidského genomu (hg19)pomocí softwarového balíčku Burrows-Wheeler Aligner (BWA) pro více vidění (Li a Durbin, 2009). Po zarovnání byly výstupní soubory použity k provedení sekvenačního pokrytí a hloubkové analýzy cílové oblasti, jednonukleotidových variant (SNVs) a indel volání, použili jsme software GATK k detekci SNV a indels (McKenna et al., 2010), všechny SNV a indely byly filtrovány a odhadovány prostřednictvím více databází, včetně databáze Jednonukleotidového polymorfismu Národního centra pro biotechnologické informace (NCBI) (dbSNP), HapMap, datasetu projektu 1000 genomů a databáze 100 čínských zdravých dospělých. K predikci účinku variant jsme použili Condel, SIFT, PolyPhen-2, LRT, mutation Taster a PhyloP. Patogenní varianty jsou hodnoceny podle protokolu vydaného Americkou vysokou školou lékařské genetiky a genomiky (ACMG) (Richards et al., 2015). Databáze lidských genových mutací (Hgmd) byla použita k screeningu mutací. Všechny potenciální patogenní varianty byly validovány pomocí sangerových sekvenačních metod.
extrakce RNA, PCR a sekvenování
mononukleární buňky periferní krve (Pmbc) a mononukleární buňky pupečníkové krve (Cbmc) byly izolovány separací gradientu hustoty Ficollu. Celková RNA byla extrahována z Pmbc a Cbmc pomocí Trizolu (Takara, Japonsko). Extrahované celkové RNA byly reverzní transkribovány pomocí RT Kit (Takara, Japonsko). PCR byla provedena pomocí GoldStar Best Master Mix (CWBIO, Peking). Sekvence primerů jsou uvedeny: L1CAM-DNA-5F, CCCACCCGTCCTTTCCTA; L1CAM-DNA-5R, CGCTCGTCCTGCTTGATGT; L1CAM-mRNA-4-6-F, GGTGTCCACTTCAAACCCAA; a L1CAM-mRNA-4-6-R, GCGGCTTCCTGTCAATCA. Sangerovo sekvenování bylo provedeno analyzátorem DNA ABI 3130.
výsledky
28letá zdravá žena byla předána na naši kliniku po čtyřech dobrovolných ukončení těhotenství v důsledku fetálního hydrocefalu. Všechny plody byly samci (obrázek 1A). Zdálo se, že rodinný rodokmen ukazuje XLH. Ve 26. týdnu těhotenství už byla v pátém těhotenství. Fetální ventrikulomegalie byla detekována ultrazvukovým vyšetřením plodu a MRI, které důsledně prokázaly přítomnost hydrocefalu. Ukázali, že bilaterální mozková komora a třetí komora byly zjevně rozšířeny a v intracerebrální a agenezi corpus callosum byl závažný hydrocefalus (obrázek 1B).
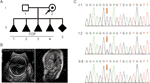
Obrázek 1 (A) rodokmen rodiny. TOP, ukončení těhotenství. B) zobrazovací vyšetření plodu. Fetální ultrazvukové vyšetření a MRI plodu ukázaly, že u plodu byl těžký hydrocefalus. (C) sekvenční analýza genomové DNA od členů rodiny. Genotypy L1CAM byly divokého typu, c. 453G > T Het, a c. 453G > T Hom, v I:1 (manžel), I:2 (těhotná žena) a II:5 (plod). Mutace je označena červenými šipkami.
abychom prozkoumali možnou genetickou příčinu, provedli jsme analýzu karyotypu a SNP pole k analýze odběru vzorků krve plodu a nenalezli žádné pozitivní nálezy. Výsledek choroidální neovaskularizace (CNV) byl uložen v genové expresi Omnibus (GEO); přístupové číslo je GSE133063, jak je připojeno níže (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
potom jsme zjistili plod Wesem. Analytická strategie pro nalezení pravděpodobné identifikace patogenních variant byla znázorněna na obrázku S1. Seznam variant (tabulka S1) byl získán screeningem variantních frekvencí, stavu mutace a režimu dědičnosti. Vezmeme-li v úvahu geny spojené s hydrocefalem (HP:0000238, http://compbio.charite.de/hpoweb/showterm?id=HP:0000238#id=HP_0000238) (tabulka S2), nedošlo k žádné další významné mutaci kromě tiché mutace c. 453G > T v exonu 5 genu L1CAM (NM_000425. 3). c. 453G > T nebyl hlášen v HGMD a Clinvaru a nebyl nalezen v databázích dbSNP, gnomAD a dalších datových sadách. Podle norem a pokynů ACMG (Richards et al., 2015), dosud nedosáhlo kritéria „patogenní“ nebo „pravděpodobně patogenní“, ale neexistovaly žádné další potenciální mutace; neměli jsme jinou možnost, než provést další analýzu nalezené tiché mutace.
podle tradičního myšlení došlo k této substituci báze ve třetí bázi v kodonu 151, který kóduje glycin, čímž se vytvořila neutrální mutace (s. Gly151 = ). Tato varianta byla potvrzena v DNA extrahované z fetální pupečníkové krve a periferní krve u páru sangerovým sekvenováním (obrázek 1C). Žena nesla heterozygotní mutaci a její manžel byl genotyp divokého typu.
s ochutnávkou mutací (http://www.mutationtaster.org/) bylo c. 453G > T hodnoceno jako “ onemocnění způsobující.“Ukázalo se, že proteinové vlastnosti mohou být ovlivněny a místo spojení může být změněno; byli jsme zvědaví na potenciální sestřihové účinky funkce L1CAM v této tiché mutaci. Tichá mutace byla testována pomocí následujících online softwarových produktů: NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) a NNSplice (http://www.fruitfly.org/seq_tools/splice.html); předpokládalo se, že v L1CAM c bude také vytvořeno potenciální spojení 5′.453G > t mutace pomocí softwarových produktů (obrázek S2). Výsledky ukázaly, že tato tichá mutace vytvořila potenciální 5′ spojovací místo 72 bp proti proudu od normálního spojovacího místa exon 6/intron 6 (obrázek 2A). Pokud tomu tak je, najdeme změnu délky l1cam messenger RNA (mRNA) mezi I:2 a II:5 (obrázek 2A). RT-PCR byla provedena pomocí primerů určených k amplifikaci exonů 4-6 v mRNA L1CAM. Výsledky skutečně ukázaly krátké pásmo zkrácení ve fetální cDNA PCR (II: 5), zatímco pásmo zesílené z l1cam mRNA obsahovalo očekávané dlouhé pásmo v manželské cDNA PCR (I:1) a dlouhé/krátké pásy u těhotné ženy cDNA PCR (I: 2) (Obrázek 2B). Přímé sekvenování amplifikovaného fragmentu ukázalo, že delece zahrnovala posledních 72 bp exonu 5 v mužské fetální cDNA (žena byla nosičem) (obrázek 2C). Máme zásadní patogenní důkazy.
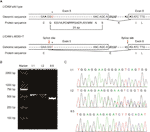
Obrázek 2 (a) schematické znázornění organizace exon 5, intron 6 a exon 6 V L1CAM. B) RT-PCR analýza exonů 5 a 6 cDNA L1CAM z mononukleárních buněk periferní krve (Pmbc) a mononukleárních buněk pupečníkové krve (Cbmc). Agarózová gelová elektroforéza RT-PCR produktů generovaných z I:1 (manžel), I:2 (těhotná žena) a II:5 (plod). C) sekvenční analýza produktu RT-PCR z Pmbc páru a Cbmc plodu.
tato tichá mutace vedla k 24 aminokyselinám proteinu L1CAM (rezidua 151-174); Lys (K) byl substituován Glu (E) v kodonu 175 (obrázek 2A). Došlo k zarovnání několika proteinových sekvencí L1CAM u několika druhů a zachování chybějících aminokyselin v L1CAM u savců: Homo sapiens, Pan troglodytes, Bos taurus, musculus a Rattus norvegicus (obrázek 3A). Divoký typ A c. 453G > t sestřih mutace l1cam proteiny byly předpovězeny softwarem Cphmodels-3.2 Server (http://www.cbs.dtu.dk/services/CPHmodels/) (obrázek 3B). Imunoglobulin-like (ig-like) doména 2 (rezidua 134-230) divokého typu a sestřih mutace l1cam proteiny je znázorněno na obrázku 3C. L1CAM c. 453G > t sestřih mutace změnil strukturu proteinu, zejména ig-like doména 2

obrázek 3 (A) zarovnání více sekvencí proteinu L1CAM napříč druhy. L1cam c.453G > T vedlo k tomu, že v konzervované aminokyselinové oblasti u různých druhů chybělo 24 aminokyselin proteinu L1CAM (rezidua 151-174). Černý sloupec zobrazuje chybějící aminokyseliny. (B) struktury divokého typu A c. 453G > t sestřih mutace l1cam protein, jak předpovídal software Cphmodels-3.2 Server. (C) struktury imunoglobulin-like (ig-like) domény 2 (rezidua 134-230) divokého typu a sestřih mutace l1cam proteinu.
diskuse
tiché mutace byly často detekovány WES, ale nebyla věnována dostatečná pozornost, což vedlo k vynechání DMs. V této studii, zaměstnali jsme Wese, aby prozkoumal genetickou příčinu čínské rodiny s hydrocefalem, ale našel pouze novou tichou mutaci v L1CAM, což nás přinutilo provést další analýzu. Naštěstí jsme dokázali, že tichá mutace vytvořila nový 5′ splice web a byla DM.
mutace v L1CAM mohou způsobit onemocnění L1 spojené s X, ale klinické příznaky jsou variabilní; mutace produkují neočekávané fenotypy. Ve studii, pět trpících plodů jsou všichni muži, což je v souladu se vzorem dědičnosti. Ultrazvukové vyšetření plodu a MRI vykazují typické onemocnění L1, včetně XLH a ageneze corpus callosum. Zlepšuje naše chápání korelace genotyp-fenotyp l1cam.
L1CAM c. 453G > T (s. Gly151 =) se původně předpokládalo, že nemá žádný vliv na proteinovou sekvenci. Ale jiné tiché mutace, c. 924C > T (s. Gly308 =) a c. 645C > T (s. Gly215 =), v genu L1CAM byly hlášeny jako DMs (Du et al ., 1998; Vos et al., 2010). D.924c > t mutace vedla k aktivaci nového spojovacího místa 69 bp 5′ na normální spojovací místo dárce exon 8 / intron 8 a bylo prohlášeno za místo „způsobující onemocnění“ pro hydrocefalus (Du et al ., 1998). Pro c. 645C > T v L1CAM bylo 51 bp odstraněno aktivací nového spojení dárce exon 6 / intron 6 (Vos et al., 2010). Naše současná studie byla podobná; mutace c. 453G > t vytvořila potenciální 5′ spojovací místo proti proudu od normálního spojovacího místa exon 5/intron 5. Všechny tyto tiché mutace vytvořily nová místa pro spojení dárců, což vedlo k vynechání exonu. Připomnělo nám, abychom věnovali velkou pozornost těmto tichým mutacím, které mohou ovlivnit sestřih proteinů.
jako transmembránový glykoprotein a člen imunoglobulinové nadčeledi buněčných adhezních molekul může protein L1CAM interagovat na buněčném povrchu s řadou různých glykoproteinů a homofilní vazba je pravděpodobně jeho hlavním způsobem interakce (Wei a Ryu, 2012). Studie krystalové struktury domén podobných ig 1-4 v neurofascinu naznačují, že mnoho patologických mutací L1 ovlivňuje konzervované aminokyselinové zbytky v těchto doménách a interferuje s homofilními interakcemi (Liu et al ., 2011), zejména jak bylo ověřeno funkčním výzkumem ig-like domény 2 (Zhao et al., 1998). V naší studii jsme spekulovali, že L1CAM c. 453G > t změnil ig-like doménu 2 v extracelulární části proteinu L1CAM, což vedlo k abnormální extracelulární interakci, selhání zahájení po signální dráze. Další studie věnovaná hmotnostní spektrometrii této varianty L1CAM by konkrétně objasnila, jaký molekulární soubor se v buňce produkuje.
stručně řečeno, prostřednictvím WES jsme hlásili novou tichou mutaci c. 453G > T v L1CAM, která produkuje 5 ‚ spojovací místo zodpovědné za hydrocefalus. Předpokládalo se, že tato abnormální proteinová varianta změní doménu 2 podobnou Ig, což by mohlo ovlivnit homofilní vazbu l1cam proteinu. Kromě toho jsme provedli prenatální genetickou diagnostiku těhotné ženy, která hlásila pět nepřetržitých těhotenství s hydrocefalem. Mezitím, navrhl některé tiché mutace detekované ve WES by neměly být ignorovány; sestřihové předpovědi těchto mutací byly nezbytné. Poskytla nový genetický základ pro prenatální diagnostiku a preimplantační prenatální diagnostiku hydrocefalu.
dostupnost dat
veřejně dostupné datové sady byly analyzovány v této studii. Tato data lze nalézt zde: GSE133063 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
etické prohlášení
studie zahrnující lidské účastníky byly přezkoumány a schváleny revizní komisí ženské nemocnice, lékařské fakulty, Zhejiang University v Číně. Písemný informovaný souhlas s účastí v této studii poskytl zákonný zástupce účastníků/nejbližší příbuzní. Písemný informovaný souhlas byl získán od fyzické osoby(osob) a nezletilého (nezletilých) zákonného zástupce / nejbližšího příbuzného pro zveřejnění potenciálně identifikovatelných obrázků nebo údajů obsažených v tomto článku.
autorské příspěvky
YS, YLi, MC, YLu, YQ a YY provedly experimenty. YS připravil čísla. MC a YLi analyzovali data WES. YLU a YQ provedly analýzu karyotypu a SNP pole. YY rekrutoval vzorky. Hl a FL poskytly zobrazovací vyšetření. YS a MD napsali rukopis. Všichni autoři přečetli a schválili konečný rukopis.
financování
tato studie byla podpořena Národní přírodovědnou nadací Číny (Grant č. 81801441), klíčovým výzkumným a vývojovým programem provincie Zhejiang (Grant č. 2019C03025), Národní klíčový program výzkumu a vývoje Číny (Grant č. 2016YFC1000703) a lékařská vědeckovýzkumná nadace provincie Zhejiang (Grant č. 2014KYA246).
Prohlášení o střetu zájmů
autoři prohlašují, že výzkum byl proveden bez jakýchkoli obchodních nebo finančních vztahů, které by mohly být vykládány jako potenciální střet zájmů.
poděkování
Děkujeme pacientům zařazeným do tohoto výzkumu. Děkujeme Dr .. Jiong Gao (BGI Genomics, BGI-Shenzhen, Shenzhen 518083, Čína) za jeho pomoc při přípravě tohoto rukopisu.
doplňkový materiál
doplňkový materiál k tomuto článku lze nalézt online na adrese: https://www.frontiersin.org/articles/10.3389/fgene.2019.00817/full#supplementary-material
obrázek S1 / Analytická strategie pro nalezení pravděpodobné identifikace patogenních variant pomocí WES.
obrázek S2 | Donor splice sites predicated by NetGene2 and NNSplice.
tabulka S1 / seznam variant prostřednictvím screeningu variant frekvencí, stavu mutace a režimu dědičnosti.
tabulka S2 | seznam genů spojených s hydrocefalem (Export pro HP: 0000238).
Du, Y.Z., Dickerson, C., Aylsworth, A. S., Schwartz, C. E. (1998). Tichá mutace, C924T (G308G), v genu L1CAM vede k X vázanému hydrocefalu (HSAS). J.Med. Genete. 35 (6), 456–462. doi: 10.1136 / jmg.35.6.456
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Itoh, K., Fushiki, S. (2015). Úloha L1cam v myší kortikogenezi a patogenezi hydrocefalu. Patol. Int. 65 (2), 58–66. doi: 10.1111 / kolík.12245
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Li, H., Durbin, R. (2009). Rychlé a přesné zarovnání krátkého čtení s transformací Burrows-Wheeler. Bioinformatika 25 (14), 1754-1760. doi: 10.1093 / bioinformatika / btp324
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Liu, h., Focia, P. J., He, X. (2011). Homofilní adhezní mechanismus neurofascinu, člena rodiny L1 adhezních molekul neurálních buněk. J.Biol. Cheme. 286 (1), 797–805. doi: 10.1074 / jbc.M110.180281
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
McKenna, a., Hanna, m., Banks, e., Sivachenko, a., Cibulskis, k., Kernytsky, A., et al. (2010). Soubor nástrojů pro analýzu genomu: rámec MapReduce pro analýzu dat sekvenování DNA nové generace. Genome Res.20 (9), 1297-1303. doi: 10.1101 / gr.107524.110
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Richards, s., Aziz, N., Bale, s., Bick, D., Das, s., Gastier-Foster, J., et al. (2015). Standardy a pokyny pro interpretaci sekvenčních variant: společné konsensuální doporučení Americké vysoké školy lékařské genetiky a genomiky a Asociace pro molekulární patologii. Genete. Med. 17 (5), 405–424. doi: 10.1038 / gim.2015.30
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
vos, y. J., de Walle, h. E. K., Bos, K. K., Stegeman, J. a., Berge, A. M., Bruining, M., et al. (2010). Korelace genotypu a fenotypu u syndromu L1: průvodce genetickým poradenstvím a analýzou mutací. J.Med. Genete. 47 (3), 169–175. doi: 10.1136 / jmg.2009.071688
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Wei, C. H., Ryu, S. E. (2012). Homofilní interakce rodiny L1 buněčných adhezních molekul. Expo. Molo. Med. 44 (7), 413–423. doi: 10.3858 / emm.2012.44.7.050
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Weller, S., Gartner, J. (2001). Genetické a klinické aspekty hydrocefalu spojeného s X (choroba L1): mutace v genu L1CAM. Hučení. Mutate. 18 (1), 1–12. doi: 10.1002 / humu.1144
PubMed Abstract | CrossRef Full Text | Google Scholar
Zhao, X., Yip, P. M., Siu, C. H. (1998). Identification of a homophilic binding site in immunoglobulin-like domain 2 of the cell adhesion molecule L1. J. Neurochem. 71 (3), 960–971 doi: 10.1046/j.1471-4159.1998.71030960.x.
PubMed Abstract | CrossRef Full Text | Google Scholar