teknologien til DNA-sekventering blev udviklet tilbage i 1977 takket være Frederick Sanger. Det tog lidt længere tid, før det var muligt at sekvensere et komplet genom. Dette skyldes, at vi havde brug for en passende matematisk model og massiv beregningskraft til at samle millioner eller milliarder små læsninger til et større komplet genom. Dagens computerkraft og programmel er den største forskel mellem, hvad der plejede at tage mange års arbejde i begyndelsen af 2000 ‘ erne, og hvad der kun tager et par timer i dag. Algoritmen du valgte at gøre dette er den” hellige gral ” af samlingsteknologien. Disse algoritmer inkorporerer en af de mest berømte variabler, der er kendt i matematiske modeller, k-mer.
oprindelsen af k-mer og den matematiske model, der omgiver den, kommer fra en 1735 Svensk matematiker Leonhard Euler, der er kendt som far til den matematiske funktion. En hollandsk matematiker Nicolaas De Bruijn tilpassede Eulers ideer til at finde en cyklisk sekvens af bogstaver taget fra et givet alfabet, for hvilket ethvert muligt ord af en bestemt længde vises som en streng af på hinanden følgende tegn i den cykliske sekvens nøjagtigt en gang.
de Bruijns algoritme blev tilpasset af molekylærbiologer, som mange år senere stod over for et tilsvarende problem: hvordan man samler DNA-sekvenser. Således bruger forskere over hele verden nu de Bruijn-grafen og variablen k.
anvendelse af k-mers til samling af DNA-sekvenser
med få ord involverer de novo genomsamling at forbinde på hinanden følgende små DNA-aflæsninger og ende med større sekvenser. For at generere en De Bruijn-graf (se figuren nedenfor) skal nukleotiderne ved kanten af hver læsning overlappe kanten af en anden (og så videre). Det endelige mål er at skabe et sammenhængende toppunkt, som (potentielt) vil resultere i store DNA-fragmenter.
du skal fragmentere dine læsninger i k-mers, som er et specifikt antal nukleotider, der overlapper hinanden. K-mer giver dig mulighed for at generere en unik sekvens fra mange små. Hver unik k-mer-sekvens identificeres, og ekstra kopier elimineres. Dette aspekt af k-mers giver dig mulighed for at overvinde en af ulemperne ved næste generations sekventering — at få læsninger, der repræsenterer genomiske regioner med forskellige frekvenser (dvs.at få en masse små læsninger fra en region). Brugen af k-mers eliminerer sekvenser gentaget mere end en gang på grund af ulige sekvensdækning. Husk dog, at en lav k-mer-størrelse vil øge chancerne for, at nukleotider overlapper hinanden, mens en større værdi vil mindske dem.
nutidens de novo–samlingsteknologi er mere effektiv, når du bruger biblioteker med store læsninger (dvs.1.000-10.000 bps) kombineret med mindre (100-200 bps). Programmer kan bruge K værdi og k-mers til at samle korte læser. Disse kan derefter inkorporeres og verificeres af større for at ende i mere nøjagtige contigs.
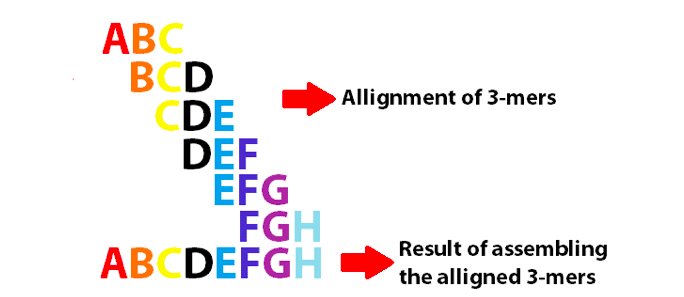
eksempel på en de Bruijn-graf ved hjælp af 3-mers til at samle de 8 første bogstaver i det engelske alfabet. Bemærk, at disse 3-mers overlapper som k-1.
jo mere du ved, jo mere kan du opnå i DNA-samling
der er specifikke tip, du skal overveje, før du anvender de Bruijn-grafer i din samlingsmetode og vælger den mest passende k-mer-størrelse. Ved at udnytte disse kan du generere bedre resultater.
- først og fremmest, og måske vigtigst, er at bruge mange forskellige k-mers i din samling. Du skal derefter evaluere dine resultater og vælge den bedste(E). Glem aldrig, at der næsten aldrig er en og kun en korrekt samling.
- du skal omhyggeligt håndtere fejllæsninger, før du bruger en k-mer. Hvis du ikke forsigtigt fjerner fejlene, kan resultaterne skabe en uønsket udbulning, der komplicerer din samling. Forøg tærsklen for den fejlrate, du bruger under sekvenstrimning. Du kan miste nogle sekvenser, men de, der forbliver, vil være de fineste.
- du skal håndtere omhyggeligt DNA-gentagelser. For eksempel genererer Illumina-sekventering en meget stor mængde data. Prøv først at samle en lille brøkdel af læsningerne, og brug dem alle til at få øje på forskelle. Gentagelige korte læsninger kan forstyrre din samleproces negativt.
- Kend dine data. Hvis du ikke kender størrelsen på dit forventede genom, mængden af sekventeringsdækning og antallet af læsninger, er du mere tilbøjelig til at vælge den bedste k-værdi til samling af dit genom. Du kan besøge k-mer-rådgivere, som velvet advisor fra Monash university for at få nogle råd om, hvilken værdi der synes mere passende.
brug af k-mers af forskellige længder og justering af contigs hjælper også forskere med at få øje på mutationshastigheder og udvide brugen. Selvfølgelig, manipulere de Bruijn grafer mod montage fordel er ikke et universalmiddel. Der er mange ting at overveje end en forenklet funktion til samling af genomet af en levende organisme. Dette er blot en introduktion af historien, og hvordan biologer kan bruge den mere effektivt.
- Compeau PE, Pevsner PA, Tesler G. (2011). Hvordan man anvender de Bruijn grafer til genom samling.Naturbioteknologi. 29(11):987–91.
- Aggarvala V, Voight BF. (2016). En udvidet sekvenskontekstmodel forklarer bredt variabilitet i polymorfismeniveauer på tværs af det humane genom. Naturgenetik. 48(4): 349–55.

har dette hjulpet dig? Så del venligst med dit netværk.