introduktion
L1-celleadhæsionsmolekylet (L1CAM) er et neuronalt celleadhæsionsmolekyle, der tilhører immunoglobulin superfamilien; det besidder nøglefunktioner i udviklingen af nervesystemet (Itoh og Fushiki, 2015). Mutationer i L1CAM har været relateret til K-linkede neurologiske syndromer, som opsummeres som L1-sygdomme. De er klassificeret som følger: På grund af stenose af akvedukten af Sylvius (HSAS), MASA syndrom (intellektuel handicap, afasi, blandet gang, adducerede tommelfingre), spastisk paraparesis type 1 (SP1) og K-forbundet agenese af corpus callosum (ACC) (Veller og Gartner, 2001; Itoh og Fushiki, 2015).
om 282 sygdomsfremkaldende mutationer (DMs) i L1CAM-genet er blevet rapporteret i hgmd Kurt Professional 2019.2 (https://portal.biobase-international.com/hgmd/pro/all.php). Ændringer i L1CAM-genet varieres; mutationsdataanalyser fra 282 patienter afslører 51% missense-og nonsensmutationer, 25% sletninger, 5% indsættelser og 19% splejsningsstedsændringer, men tavse mutationer i L1CAM med patogen potentiale var sjældne, og tavse mutationer blev ofte ignoreret, især ved påvisning af heleksomsekvensering.
i denne undersøgelse screenede vi føtal DNA fra en kinesisk gravid kvinde, der har rapporteret fem kontinuerlige graviditeter med føtal hydrocephalus; vi fandt kun en ny stille mutation c.453g > T (s.Gly151 = ) i L1CAM-genet. Interessant nok indikerede vi gennem yderligere analyse, at den tavse mutation skabte en potentiel 5 ‘ splice site konsensussekvens, hvilket ville resultere i en in-frame-sletning af 72 bp fra ekson 5 og 24 aminosyrer af l1cam-proteinet.
Case præsentation
en 28-årig sund kvinde blev henvist til vores klinik efter fire frivillige ophør af graviditet på grund af føtal hydrocephalus på andre hospitaler. Alle fostre var mandlige. Da hun ankom til vores hospital (Kvindehospital, medicinsk skole, Jejiang Universitet, Jejiang, Kina), var hun allerede på sin femte graviditet ved 24 ugers drægtighed med en føtal hydrocephalus ved billedundersøgelser. For at undersøge den genetiske årsag blev føtal blodprøveudtagning udført ved 26 ugers svangerskabsalder. Konventionelle cytogenetiske undersøgelser blev udført for både føtal-og forældreprøver, og føtalprøven blev yderligere analyseret ved hjælp af single-nucleotide polymorphism (SNP) array og ves.
denne undersøgelse blev udført i overensstemmelse med anbefalingerne fra det etiske udvalg for kvinders Hospital, School of Medicine Jejiang University, og informeret samtykke blev erhvervet fra alle deltagerne i denne undersøgelse i overensstemmelse med Helsinki-erklæringen. Undersøgelsesprotokollen blev godkendt af undersøgelsesudvalget for Kvindehospitalet, School of Medicine, Jejiang University i Kina.
materialer og metoder
Karyotype og SNP Array
karyotyperne af føtal ledningsblod og perifert ledningsblod blev bestemt ved konventionel karyotyping af mindst 30 blodlymfocytter, som blev arresteret ved metafase af colchiciner. G-banding karyotyper af dyrkede celler blev udført på 320-400 båndniveau med en opløsning på omkring 10 Mb. 2.600.000 markører inklusive 750.000 SNP-prober og 1.900.000 ikke-polymorfism-prober til omfattende helgenomdækning. Data blev analyseret ved hjælp af Kromosomanalyseprogrammet (ChAS) (Affymetrics, Santa Clara, CA) baseret på grch37/hg19-samlingen. Rapporteringstærsklen for kopinummerresultatet blev sat til 500 kb med et markørantal på kr50 for gevinster og til 200 kb med et markørantal på kr50 for tab.
hel-Eksom sekventering
hoveddelen af VES blev leveret af Beijing Genomics Institute. Genomisk DNA blev ekstraheret af et DNeasy Blodsæt og blev derefter fragmenteret af Covaris LE220 (Massachusetts, USA) for at generere et parret bibliotek (200-250 bp). Alle forstærkede biblioteker blev udført på bgisek-500-platformen, enkeltstrenget DNA blev blandet med mgieasy rep DNA Library Kit V1 (BGI, Shenjen, Kina) og derefter sekventeret ved hjælp af 100sr-kemi med bgisek-500Rs sekventeringssæt med høj kapacitet (BGI, Shenjen, Kina).
rene aflæsninger (med en længde på 90 bp) afledt af målrettet sekventering og filtrering blev derefter justeret til human genome reference (hg19) ved hjælp af Multi-Vision programpakke til Gravhjul (Li og Durbin, 2009). Efter justering blev outputfilerne brugt til at udføre sekventeringsdækning og Dybdeanalyse af målområdet, enkeltnukleotidvarianter (SNV ‘er) og Indel-opkald, vi brugte gatk-programmet til at detektere SNV’ er og indels (McKenna et al., 2010), blev alle SNV ‘er og Indel’ er filtreret og estimeret via flere databaser, herunder National Center for Biotechnology Information (NCBI) single-Nucleotide Polymorphism Database (dbSNP), HapMap, 1000 genomer projekt datasæt og database med 100 kinesiske raske voksne. Vi brugte Condel, SIFT, PolyPhen-2, LRT, Mutationstaster og PhyloP til at forudsige effekten af varianter. Patogene varianter vurderes i henhold til protokollen udstedt af American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015). Den humane Genmutationsdatabase (HGMD) blev brugt til at screene mutationer. Alle potentielle patogene varianter blev valideret ved hjælp af Sanger-sekventeringsmetoder.
RNA-ekstraktion, PCR og sekventering
mononukleære celler i perifert blod (Pmbc ‘er) og mononukleære celler i navlestrengsblod (Cbmc’ er) blev isoleret ved ficoll densitetsgradientseparation. Det samlede RNA blev ekstraheret fra Pmbc ‘er og Cbmc’ er ved hjælp af Trisol (Takara, Japan). Ekstraherede samlede RNA ‘ er blev omvendt transkriberet ved hjælp af RT Kit (Takara, Japan). PCR blev udført ved hjælp af GoldStar Best Master blanding (Cbio, Beijing). Primersekvenser er angivet: L1CAM-DNA-5F, CCCACCCGTCCTTTTCCTA; L1CAM-DNA-5R, CGCTCGTCCTGCTGATGT; L1CAM-mRNA-4-6-F, GGTGTCCACTTCAAACCCAA; og L1CAM-mRNA-4-6-R, GCGGCTCCTGTCAATCA. Sanger-sekventering blev udført af en ABI 3130 DNA-analysator.
resultater
en 28-årig sund kvinde blev henvist til vores klinik efter fire frivillige ophør af graviditet på grund af føtal hydrocephalus. Alle fostre var mandlige (figur 1a). Den familiære stamtavle syntes at vise HLH. Hun var allerede på her femte graviditet ved 26 ugers svangerskab. Føtal ventrikulomegali blev påvist ved føtal ultralydscanning og MR, som konsekvent viste tilstedeværelsen af hydrocephalus. De viste, at den bilaterale cerebrale ventrikel og den tredje ventrikel åbenbart var udvidet, og der var svær hydrocephalus i intracerebral og agenese af corpus callosum (figur 1b).
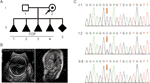
Figur 1 (A) stamtavle af familien. TOP, ophør af graviditet. (B) billeddannelsesundersøgelser af fosteret. Føtal ultralydsscanning og føtal MR viste, at der var alvorlig hydrocephalus hos fosteret. (C) sekvensanalyse af genomisk DNA fra familiemedlemmer. Genotyperne af L1CAM var vildtype, C. 453g > T Het, og c. 453g > t Hom, hos I:1 (mand), i:2 (gravid kvinde) og II:5 (Foster). Mutationen er angivet med de røde pile.
for at undersøge den mulige genetiske årsag udførte vi karyotypeanalyse og SNP-array for at analysere føtal blodprøveudtagning og fandt ingen positive fund. Choroidal neovaskularisering (CNV) resultat er blevet deponeret i genekspression Omnibus (GEO); tiltrædelsesnummeret er GSE133063, som vedlagt nedenfor (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
derefter opdagede vi fosteret af VES. Den analytiske strategi til at finde sandsynlig patogen variantidentifikation blev vist i figur S1. En liste over varianter (tabel S1) blev opnået gennem screening af variantfrekvenser, mutationsstatus og arvstilstand. Under hensyntagen til de hydrocephalus-associerede gener (HP:0000238, http://compbio.charite.de/hpoweb/showterm?id=HP:0000238#id=HP_0000238) (tabel S2) var der ingen yderligere bemærkelsesværdig mutation bortset fra den tavse mutation af c.453g > T i ekson 5 af L1CAM-genet (NM_000425.3). c. 453g > T blev ikke rapporteret i HGMD og ClinVar og blev ikke fundet i dbSNP, gnomAD og andre datasæt. I henhold til standarderne og retningslinjerne fra ACMG (Richards et al., 2015), havde den endnu ikke nået kriteriet om “patogen” eller “sandsynlig patogen”, men der var ingen andre potentielle mutationer; vi havde intet andet valg end at foretage en yderligere analyse af den fundne tavse mutation.
ifølge traditionel tænkning forekom denne basesubstitution i den tredje base i codon 151, som koder for en glycin og derved skaber en neutral mutation (s.Gly151 = ). Denne variant blev bekræftet i DNA ekstraheret fra føtal ledningsblod og perifert blod i parret ved Sanger-sekventering (figur 1C). Kvinden bar den heterosygøse mutation, og hendes mand var en vildtype genotype.
med Mutationstaster (http://www.mutationtaster.org/), C.453g > T blev scoret som “sygdomsfremkaldende.”Det viste, at proteinfunktioner kan blive påvirket, og splejsningsstedet kan ændres; vi var nysgerrige efter de potentielle splejsningseffekter af l1cam-funktionen i denne tavse mutation. Den tavse mutation blev testet ved hjælp af følgende Onlineprogrammer: NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) og NNSplice (http://www.fruitfly.org/seq_tools/splice.html); det 5′ potentielle splejsningssted blev også forudsagt at blive oprettet i L1CAM c.453g > t mutation ved hjælp af programmelprodukterne (figur S2). Resultaterne viste, at denne tavse mutation skabte et potentielt 5 ‘ splejsningssted 72 bp opstrøms fra det normale ekson 6 / intron 6 splejsningssted (figur 2a). Hvis dette er tilfældet, kan vi finde længdeændringen af L1CAM messenger RNA (mRNA) mellem I:2 og II:5 (figur 2a). RT-PCR blev udført under anvendelse af primere designet til at amplificere eksoner 4-6 i l1cam mRNA. Faktisk viste resultaterne et kort trunkeringsbånd i føtal cDNA PCR (II: 5), mens båndet forstærket fra L1CAM mRNA indeholdt det forventede lange bånd i mand cDNA PCR (I:1) og lange/korte bånd i den gravide kvinde cDNA PCR (I:2) (Figur 2b). Direkte sekventering af det forstærkede fragment viste, at sletningen involverede de sidste 72 bp af ekson 5 i mandlig føtal cDNA (kvinden var en bærer) (figur 2C). Vi har de afgørende patogene beviser.
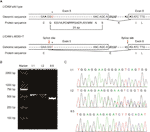
figur 2 (a) skematisk repræsentation af ekson 5, intron 6og ekson 6 organisation i L1CAM. B) RT-PCR-analyse af eksoner 5 og 6 i l1cam cDNA fra mononukleære celler i perifert blod (Pmbc ‘er) og mononukleære celler i navlestrengsblod (Cbmc’ er). Agarosegelelektroforese af RT-PCR-produkter genereret fra I: 1 (mand), i:2 (gravid kvinde) og II:5 (Foster). C) sekvensanalyse af RT-PCR-produktet fra pmbc ‘er hos parret og Cbmc’ er hos fosteret.
denne tavse mutation resulterede i 24 aminosyrer af L1CAM-protein (rester 151-174); Lys (K) blev substitueret med Glu (E) ved codon 175 (figur 2a). Der var tilpasning af flere l1cam-proteinsekvenser på tværs af flere arter og bevarelse af de manglende aminosyrer i L1CAM på tværs af pattedyr: Homo sapiens, Pan troglodytes, Bos taurus, Mus musculus og Rattus norvegicus (figur 3a). Vildtype og c.453g > T splejsningsmutation l1cam-proteiner blev forudsagt af cphmodels-3.2-serveren (http://www.cbs.dtu.dk/services/CPHmodels/) (figur 3b). Immunoglobulinlignende (ig-lignende) domæne 2 (rester 134-230) af vildtype-og splejsningsmutation L1CAM-proteiner er vist i figur 3C. L1CAM c. 453G > T splejsningsmutation ændrede proteinstrukturen, især det Ig – lignende domæne 2

figur 3 (a) tilpasning af flere l1cam-proteinsekvenser på tværs af arter. L1cam c.453G > t resulterede i, at 24 aminosyrer af L1CAM-protein (rester 151-174) manglede i det konserverede aminosyreområde i forskellige arter. Den sorte søjle viser de manglende aminosyrer. (B) strukturer af vildtype og c.453g > T splejsning mutation L1CAM protein som forudsagt af programmet Cphmodels-3.2 Server. C) strukturerne af immunglobulinlignende (ig-lignende) domæne 2 (restkoncentrationer 134-230) af vildtype-og splejsningsmutation l1cam-protein.
Diskussion
tavse mutationer blev ofte påvist af VES, men utilstrækkelig opmærksomhed er blevet betalt, hvilket fører til udeladelse af DMs. I denne undersøgelse anvendte vi ves til at undersøge den genetiske årsag til en kinesisk familie med hydrocephalus, men fandt kun en ny stille mutation i L1CAM, som tvang os til at foretage en yderligere analyse. Heldigvis beviste vi, at den tavse mutation skabte et nyt 5′ splejsningssted og var en DM.
mutationer i L1CAM kan forårsage en KSB-forbundet L1-sygdom, men kliniske symptomer er variable; mutationer producerer uventede fænotyper. I undersøgelsen er de fem lidende fostre alle mænd, hvilket er i overensstemmelse med et arvemønster. Den føtale ultralydsscanning og MR viser en typisk L1-sygdom, herunder HLH og agenese af corpus callosum. Det forbedrer vores forståelse af genotype-fænotype-korrelationen af L1CAM.
L1CAM c.453g > T (s.Gly151 = ) blev oprindeligt antaget at have nogen effekt på proteinsekvensen. Men andre tavse mutationer, c.924c > T (s.Gly308 = ) og c.645c > T (s.Gly215 = ), i L1CAM-genet er rapporteret at være DMs (du et al., 1998; Vos et al., 2010). C.924c > t-mutation resulterede i aktivering af et nyt splejsningssted 69 bp 5′ til det normale ekson 8/intron 8 donorsplejsningssted, og det er blevet erklæret som et “sygdomsfremkaldende” sted for hydrocephalus (Du et al., 1998). For c. 645C > T i L1CAM blev 51 bp slettet med aktiveringen af en ny ekson 6/intron 6 donorsplitning (Vos et al., 2010). Vores nuværende undersøgelse var ens; mutationen af c.453g > t skabte et potentielt 5′ splejsningssted opstrøms fra det normale ekson 5/intron 5 splejsningssted. Alle disse tavse mutationer skabte nye donorsplitningssteder, hvilket resulterede i, at eksonen blev sprunget over. Det mindede os om at være meget opmærksomme på disse tavse mutationer, som kan påvirke splejsning af proteiner.
som et transmembranglycoprotein og et medlem af immunoglobulins superfamilien af celleadhæsionsmolekyler kan l1cam-proteinet interagere på celleoverfladen med et antal forskellige glycoproteiner, og homofil binding er sandsynligvis dens vigtigste interaktionsmåde (Vei og Ryu, 2012). Undersøgelserne af krystalstrukturen af IG-lignende domæner 1-4 i neurofascin antydede, at mange patologiske L1-mutationer påvirker konserverede aminosyrerester inden for disse domæner og interfererer med homofile interaktioner (Liu et al., 2011), især som verificeret af funktionsforskning af Ig-lignende domæne 2 (Jhao et al., 1998). I vores undersøgelse spekulerede vi i, at L1CAM c.453g > t ændrede Ig-lignende domæne 2 i den ekstracellulære del af l1cam-proteinet, hvilket førte til den unormale ekstracellulære interaktion, idet den ikke begyndte at starte nedstrøms signalvejen. En yderligere undersøgelse dedikeret til massespektrometri af denne L1CAM-variant ville præcisere specifikt, hvilket molekylært ensemble der produceres i cellen.
sammenfattende rapporterede vi gennem ves en ny stille mutation c. 453g > T I L1CAM, der producerer et 5′ splejsningssted, der er ansvarlig for hydrocephalus. Denne unormale proteinvariant blev forudsagt at ændre Ig – lignende domæne 2, hvilket kan påvirke l1cam-proteinhomofil binding. Derudover udførte vi prænatal genetisk diagnose for den gravide kvinde, der rapporterede fem kontinuerlige graviditeter med hydrocephalus. I mellemtiden foreslog det, at nogle tavse mutationer, der blev påvist i ves, ikke skulle ignoreres; splejsning af forudsigelser af disse mutationer var nødvendige. Det gav et nyt genetisk grundlag for prænatal diagnose og præimplantation prænatal diagnose af hydrocephalus.
datatilgængelighed
offentligt tilgængelige datasæt blev analyseret i denne undersøgelse. Disse data kan findes her: GSE133063 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
Etikerklæring
undersøgelserne, der involverede menneskelige deltagere, blev gennemgået og godkendt af bedømmelsesudvalget for Kvindehospitalet, School of Medicine, Jejiang University i Kina. Skriftligt informeret samtykke til at deltage i denne undersøgelse blev leveret af deltagernes værge/pårørende. Der blev indhentet skriftligt informeret samtykke fra den eller de enkelte og mindreåriges værge/pårørende til offentliggørelse af potentielt identificerbare billeder eller data, der er inkluderet i denne artikel.
Forfatterbidrag
YS, YLi, mc, YLu, YK og YY udførte eksperimenter. YS forberedt tallene. MC og YLi analyserede data. YLu og YK udførte karyotype analyse og SNP array. YY rekrutterede prøver. HL og FL leverede billeddannelsesundersøgelser. YS og MD skrev manuskriptet. Alle forfattere læste og godkendte det endelige manuskript.
finansiering
denne undersøgelse blev støttet af National Natural Science Foundation of China (Grant No. 81801441), det vigtigste forsknings-og udviklingsprogram for Jejiang-provinsen (Grant No. 2019c03025), Kinas nationale Nøgleforsknings-og udviklingsprogram (Grant No. 2016yfc1000703) og det medicinske videnskabelige forskningsgrundlag i Jejiang-provinsen (Grant No. 2014kya246).
interessekonflikt Erklæring
forfatterne erklærer, at forskningen blev udført i mangel af kommercielle eller økonomiske forhold, der kunne fortolkes som en potentiel interessekonflikt.
anerkendelser
vi takker de patienter, der er tilmeldt denne forskning. Tak til DR. Jiong Gao (BGI Genomics, BGI-Shenjen, Shengen 518083, Kina) for hans hjælp under forberedelsen af dette manuskript.
supplerende materiale
det supplerende materiale til denne artikel kan findes online på: https://www.frontiersin.org/articles/10.3389/fgene.2019.00817/full#supplementary-material
figur S1 / analytisk strategi for at finde sandsynlig patogen variantidentifikation af VES.
figur S2 | Donor splejsningssteder forudsagt af NetGene2 og NNSplice.
tabel S1 / liste over varianter gennem screening af varianter frekvenser, mutationsstatus og arv tilstand.
tabel S2 / liste over hydrocephalus-associerede gener (eksport til HP:0000238).
Du, Y. S., Dickerson, C., A. S., Schvarts, C. E. (1998). En tavs mutation, C924T (G308G), i l1cam-genet resulterer i HSAs (hydrocephalus). J. Med. Genet. 35 (6), 456–462. doi: 10.1136 / jmg.35.6.456
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
Itoh, K., Fushiki, S. (2015). L1cams rolle i Murin kortikogenese og patogenesen af hydrocephalus. Pathol. Int. 65 (2), 58–66. doi: 10.1111 / pin.12245
PubMed Abstrakt | CrossRef Fuld Tekst | Google Scholar
Li, H., Durbin, R. (2009). Hurtig og præcis kortlæsningsjustering med huller-Hjultransformation. Bioinformatik 25 (14), 1754-1760. doi: 10.1093 / bioinformatik / btp324
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Liu, H., Focia, P. J., H. (2011). Homofil adhæsionsmekanisme af neurofascin, et medlem af L1-familien af neurale celleadhæsionsmolekyler. J. Biol. Chem. 286 (1), 797–805. doi: 10.1074 / jbc.M110.180281
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). Genomanalyseværktøjssættet: en MapReduce-ramme til analyse af næste generations DNA-sekventeringsdata. Genom Res. 20 (9), 1297-1303. doi: 10.1101 / gr.107524.110
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Richards, S., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standarder og retningslinjer for fortolkning af sekvensvarianter: en fælles konsensusanbefaling fra American College of Medical Genetics and Genomics og Association for Molecular Pathology. Genet. Middelhavs. 17 (5), 405–424. doi: 10.1038 / gim.2015.30
PubMed abstrakt | CrossRef Fuld tekst | Google Scholar
det er en af de mest populære måder at gøre det på. (2010). Genotype-fænotype korrelationer i L1 syndrom: en guide til genetisk rådgivning og mutationsanalyse. J. Med. Genet. 47 (3), 169–175. doi: 10.1136 / jmg.2009.071688
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
Vei, C. H., Ryu, S. E. (2012). Homofil interaktion af L1-familien af celleadhæsionsmolekyler. Eksp. Mol. Middelhavs. 44 (7), 413–423. doi: 10.3858 / emm.2012.44.7.050
PubMed Abstrakt | CrossRef Fuld Tekst | Google Scholar
(2001). Genetiske og kliniske aspekter af hydrocephalus (L1-sygdom): mutationer i l1cam-genet. Hum. Mutat. 18 (1), 1–12. doi: 10.1002 / humu.1144
PubMed Abstract | CrossRef Full Text | Google Scholar
Zhao, X., Yip, P. M., Siu, C. H. (1998). Identification of a homophilic binding site in immunoglobulin-like domain 2 of the cell adhesion molecule L1. J. Neurochem. 71 (3), 960–971 doi: 10.1046/j.1471-4159.1998.71030960.x.
PubMed Abstract | CrossRef Full Text | Google Scholar