Die Technologie zur DNA-Sequenzierung wurde bereits 1977 dank Frederick Sanger entwickelt. Es dauerte etwas länger, bis es möglich war, ein vollständiges Genom zu sequenzieren. Dies liegt daran, dass wir ein geeignetes mathematisches Modell und eine enorme Rechenleistung benötigten, um Millionen oder Milliarden kleiner Lesevorgänge zu einem größeren vollständigen Genom zusammenzusetzen. Die heutige Rechenleistung und Software sind der Hauptunterschied zwischen dem, was in den frühen 2000er Jahren jahrelang gearbeitet hat, und dem, was heute nur wenige Stunden dauert. Der Algorithmus, den Sie dafür gewählt haben, ist der „heilige Gral“ der Montagetechnologie. Diese Algorithmen enthalten eine der bekanntesten Variablen, die in mathematischen Modellen bekannt sind, den k-Mer.
Der Ursprung des k-mer und des mathematischen Modells, das ihn umgibt, stammt von einem 1735 Schweizer Mathematiker Leonhard Euler, der als Vater der mathematischen Funktion bekannt ist. Ein niederländischer Mathematiker Nicolaas de Bruijn adaptierte Eulers Ideen, um eine zyklische Folge von Buchstaben aus einem bestimmten Alphabet zu finden, für die jedes mögliche Wort einer bestimmten Länge genau einmal als Folge aufeinanderfolgender Zeichen in der zyklischen Folge erscheint.
de Bruijns Algorithmus wurde von Molekularbiologen angepasst, die viele Jahre später vor einem ähnlichen Problem standen: wie DNA-Sequenzen zusammengesetzt werden. So verwenden Wissenschaftler auf der ganzen Welt jetzt den De-Bruijn-Graphen und die Variable k.
Anwendung von k-mers auf die Assemblierung von DNA-Sequenzen
In wenigen Worten, de novo genome Assembly beinhaltet die Verbindung aufeinanderfolgender kleiner DNA-Sequenzen und endet mit größeren Sequenzen. Um einen De-Bruijn-Graph zu erzeugen (siehe Abbildung unten), müssen die Nukleotide am Rand jedes Lesevorgangs den Rand eines zweiten überlappen (und so weiter). Das endgültige Ziel besteht darin, einen aufeinanderfolgenden Scheitelpunkt zu erstellen, der (möglicherweise) zu großen DNA-Fragmenten führt.
Sie müssen Ihre Dna in k-Mere fragmentieren, bei denen es sich um eine bestimmte Anzahl von Nukleotiden handelt, die sich überlappen. Mit dem k-Mer können Sie aus vielen kleinen eine eindeutige Sequenz generieren. Jede eindeutige k-Mer-Sequenz wird identifiziert und zusätzliche Kopien werden eliminiert. Dieser Aspekt von k-mers ermöglicht es Ihnen, einen der Nachteile der Next-Generation-Sequenzierung zu überwinden – Lesevorgänge zu erhalten, die genomische Regionen mit unterschiedlichen Frequenzen darstellen (dh viele kleine Lesevorgänge aus einer Region zu erhalten). Die Verwendung von k-mers eliminiert Sequenzen, die wegen ungleicher Sequenzabdeckung mehr als einmal wiederholt werden. Beachten Sie jedoch, dass eine niedrige k-mer-Größe die Wahrscheinlichkeit erhöht, dass sich Nukleotide überlappen, während ein größerer Wert sie verringert.
Die heutige de novo Assembly-Technologie ist effizienter, wenn Sie Bibliotheken mit großen Lesevorgängen (d. H. 1.000–10.000 bps) in Kombination mit kleineren Lesevorgängen (100-200 bps) verwenden. Softwareprogramme können den k-Wert und k-mers verwenden, um kurze Lesevorgänge zusammenzustellen. Diese können dann von größeren aufgenommen und verifiziert werden, um genauere Verbindungen zu erhalten.
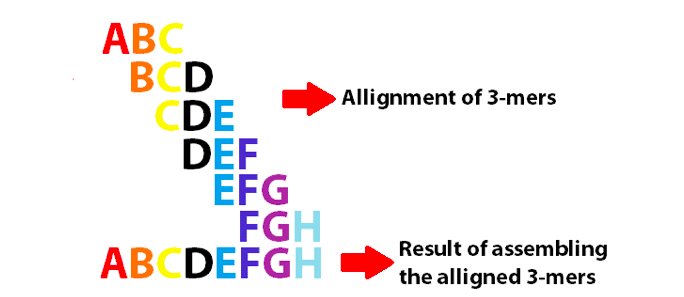
Beispiel eines de Bruijn-Graphen unter Verwendung von 3-mers zum Zusammensetzen der 8 ersten Buchstaben des englischen Alphabets. Beachten Sie, dass sich diese 3-mere als k-1 überlappen.
Je mehr Sie wissen, desto mehr können Sie bei der DNA-Montage erreichen
Es gibt bestimmte Tipps, die Sie beachten müssen, bevor Sie De Bruijn-Diagramme in Ihrer Montagemethode anwenden und die am besten geeignete k-Mer-Größe auswählen. Indem Sie diese nutzen, können Sie bessere Ergebnisse erzielen.
- Zuallererst und vielleicht am wichtigsten ist es, viele verschiedene k-mer in Ihrer Baugruppe zu verwenden. Sie sollten dann Ihre Ergebnisse bewerten und die besten auswählen. Vergessen Sie nie, dass es fast nie eine und nur eine korrekte Montage gibt.
- Sie sollten Fehlermeldungen sorgfältig behandeln, bevor Sie einen k-Mer verwenden. Wenn Sie die Fehler nicht sorgfältig entfernen, können die Ergebnisse zu einer unerwünschten Ausbuchtung führen, die Ihre Montage erschwert. Erhöhen Sie den Schwellenwert für die Fehlerrate, die Sie beim Sequenztrimmen verwenden. Sie könnten einige Sequenzen verlieren, aber diejenigen, die bleiben, werden die besten sein.
- Sie sollten vorsichtig mit DNA-Wiederholungen umgehen. Zum Beispiel erzeugt Illumina Sequencing eine sehr große Datenmenge. Versuchen Sie zuerst, einen kleinen Bruchteil der Buchstaben zusammenzusetzen, und verwenden Sie sie dann alle, um Unterschiede zu erkennen. Wiederholbare kurze Lesevorgänge können sich negativ auf Ihren Montageprozess auswirken.
- Kennen Sie Ihre Daten. Wenn Sie die Größe Ihres erwarteten Genoms, die Menge der Sequenzierungsabdeckung und die Anzahl der Lesevorgänge nicht kennen, neigen Sie eher dazu, den besten k-Wert für die Zusammenstellung Ihres Genoms zu wählen. Sie können k-Mer-Berater wie Velvet Advisor von der Monash University besuchen, um Ratschläge zu erhalten, welcher Wert besser geeignet erscheint.
Die Verwendung von k-Mers unterschiedlicher Länge und die Ausrichtung der Contigs helfen den Forschern auch, Mutationsraten zu erkennen und ihre Verwendung zu erweitern. Natürlich ist die Manipulation von De Bruijn-Diagrammen zum Nutzen der Versammlung kein Allheilmittel. Es gibt zahlreiche Dinge zu beachten, als eine vereinfachte Funktion zum Zusammenbau des Genoms eines lebenden Organismus. Dies ist nur eine Einführung in die Geschichte und wie Biologen sie effizienter nutzen können.
- Compeau PE, Pevzner PA, Tesler G. (2011). So wenden Sie de Bruijn-Diagramme auf die Genomassembly an.Natur Biotechnologie. 29(11):987–91.
- Aggarwala V, Voight BF. (2016). Ein erweitertes Sequenzkontextmodell erklärt weitgehend die Variabilität der Polymorphismusniveaus im gesamten menschlichen Genom. Naturgenetik. 48(4): 349–55.

Hat Ihnen das geholfen? Dann teilen Sie bitte mit Ihrem Netzwerk.