Hintergrund
Das intramuskuläre Myxom ist ein Weichteiltumor mesenchymalen Ursprungs unbekannter Ätiologie. Es ist selten, mit einer Inzidenz zwischen 0,1 und 0,13 pro 100.000 Personen. Der Beginn liegt in der Regel zwischen dem vierten und siebten Lebensjahrzehnt, vorwiegend bei Frauen (70%).1
Am häufigsten sind die oberen Extremitäten (Schulter und Arm), die Oberschenkel und die Gesäßmuskulatur betroffen.2 Es wurde auch in anderen Regionen wie dem Deltamuskel, dem Schulterblattbereich, der Bauchwand oder den paravertebralen Muskeln beschrieben.
Es stellt sich normalerweise als solitärer Tumor dar, obwohl es multiple Myxome gibt, die oft mit anderen Entitäten assoziiert sind und interessante Syndrome bilden.
Das Ziel unserer Studie war es, 2 klinische Fälle von intramuskulärem Myxom zu beschreiben, einzeln und mehrfach, mit Blick auf die Differentialdiagnose, Behandlung und anatomopathologische Studie. Diese Prämissen sind weitreichend, da Myxome mit malignen Läsionen verwechselt werden können oder zu einer schlechten Diagnose und unvollständigen Behandlung führen können, wenn wir uns ihrer Assoziation mit anderen Störungen, mit denen sie Syndrome bilden, nicht bewusst sind.
Klinischer Fallklinischer Fall 1
Eine 65-jährige Patientin ohne Interesse in der Anamnese mit einer linken Gesäßmasse, die sich über mehrere Jahre entwickelt hatte. Bei der Untersuchung wurde eine runde, schmerzlose, unbewegliche Masse von etwa 12 cm innerhalb der Muskulatur gefunden.
Ultraschall beschrieb eine gut definierte feste Läsion in der äußeren kaudalen Fläche des Gluteus maximus mit Merkmalen eines möglichen Sarkoms. Es gab keine Knochen- oder Gelenkbeteiligung. Die Studie wurde mit Computertomographie (CAT) abgeschlossen (Abb. 1), zeigt eine gut definierte Masse von 8 cm × 6 cm, Hypodense ohne Verstärkung nach Injektion von Kontrast in den Gluteus maximus und mit Kernspinresonanz (NMR) (Abb. 2), die diese Ergebnisse bestätigten.
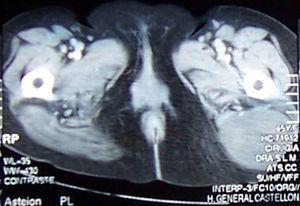
Axiale Computertomographie. Gut definierte Masse 8 cm × 6 cm, Hypodense ohne Verstärkung nach Injektion von Kontrastmittel in den Gluteus maximus.

MRT-Untersuchung. Koronale Sequenz.
Eine Narbenbiopsie wurde von der Läsion durchgeführt, die einen myxoiden Tumor ohne Anzeichen von Malignität meldete. Da jedoch die myxoide Substanz und die Bereiche Hyperzellularität und Hypervaskularität die Differentialdiagnose mit myxoiden Sarkomen erschwerten und die bildgebenden Untersuchungen eine Malignität nicht ausschlossen, wurde ein chirurgischer Eingriff beschlossen.
Die Behandlung der Wahl war die radikale Glutektomie aufgrund der Lage und Größe der Masse (Abb. 3).
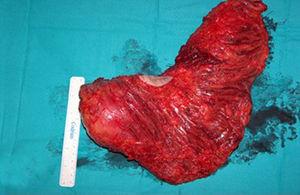
Chirurgisches Stück.
Das anatomopathologische Ergebnis des chirurgischen Stücks war ein pseudoverkapseltes intramuskuläres Myxom.
Sieben Tage nach der Operation gab es keine Komplikationen und der Patient war mobil.
Klinischer Fall 2
Eine 40-jährige Patientin wurde über mehrere Monate mit Steißbein und Schmerzen in der linken Schulter konsultiert und hatte keine persönliche Vorgeschichte von Interesse.
Bei der körperlichen Untersuchung wurden keine pathologischen Anzeichen gefunden. Es wurden einfache Hüft- und Knie-Röntgenaufnahmen durchgeführt, die normal waren. Daher wurde eine konservative Behandlung mit Analgesie verordnet.
Da die Symptome anhielten, wurde eine MRT-Untersuchung des linken Knies und des Rückens durchgeführt. Auf Höhe des Beckens wurden mehrere intramuskuläre zystische Läsionen zwischen 14 mm und 52 mm gefunden, die sich im rechten Gluteus medius und maximus befanden. Diese Läsionen befanden sich zwischen den Muskelfasern und verdrängten sie, und es gab keine Anzeichen einer Infiltration (Abb. 4). Das linke Knie zeigte einen schrägen Riss, bei dem sich der Körper mit dem hinteren Horn des inneren Meniskus verband, der auf der Koronalebene zu sehen war, ohne damit verbundene Chondropathie.
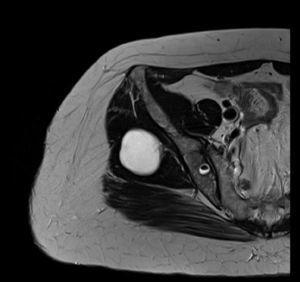
MRT-Untersuchung. Becken axial T2-gewichtet.
Die radiologischen Eigenschaften dieser Läsionen waren mit hydatidischen Zysten kompatibel, und daher wurde die Studie mit einem CAT-Scan und einer Serologie abgeschlossen. Der CAT-Scan bestätigte die intramuskulären zystischen Läsionen in den rechten Gesäßmuskeln und zeigte eine weitere kleine (16 mm) zystische Läsion mit glatten Konturen und definierten Rändern auf der Ebene des Mesenterialfetts vor dem aufsteigenden Dickdarm, die jedoch nicht mit ihm in Kontakt kam. Die Serologie war negativ für Hydatidose.
Da die Läsionen radiologisch wie hydatidische Zysten aussahen, wurde die Behandlung mit Albendazol trotz der serologischen Ergebnisse eingeleitet. Nach 6 Monaten hielten die Schmerzen der Patientin jedoch an und sie fühlte sich beim Gehen unwohl. Daher wurde ein chirurgischer Eingriff beschlossen, um die Zysten zu entfernen.
Der Patient sollte operiert werden. Sie wurde in die linke laterale Dekubitusposition gebracht und die Zysten wurden über einen Längsschnitt im rechten Oberschenkel erreicht. Die Zysten wurden intramuskulär in den Gluteus maximus und Gluteus medius eingebracht und leicht entfernt (Abb. 5). Ihr makroskopisches Aussehen deutete nicht auf hydatidische Zysten hin (Abb. 6). Während der Operation wurde ein Ultraschall durchgeführt, um die kleinsten Zysten zu lokalisieren und eine vollständige Entfernung zu erreichen. Insgesamt wurden 7 Zysten entfernt.
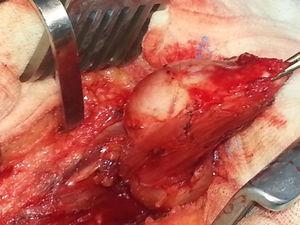
Intramuskuläre Myxoidzyste.
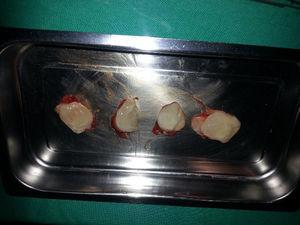
Makroskopisches Erscheinungsbild der Läsionen.
Die anatomopathologische Studie beschrieb die Läsionen als multiple intramuskuläre Myxome im Zusammenhang mit dem Mazabraud-Syndrom, wobei berücksichtigt wurde, dass die charakteristischen Knochenläsionen vor den Myxomen auftreten oder später auftreten können.
Angesichts der anatomopathologischen Ergebnisse wurde eine vollständige radiologische Studie durchgeführt, um Knochendysplasien zu lokalisieren, aber es gab keine pathologischen Befunde.
Der Patient wurde nach 5 Tagen ohne unmittelbare Komplikationen und ausreichender Funktion der rechten unteren Extremität entlassen.
Bei nachfolgenden Besuchen in der Ambulanz wurde die intraabdominale Läsion überwacht und der Patient auf nachfolgende Knochendysplasien mit periodischer Wiederholung von einfachen Röntgenstrahlen untersucht.
Diskussion
Der Begriff Myxom wurde 1863 von Virchow eingeführt, um einen mesenchymalen Tumor zu beschreiben, der histologisch der Nabelschnur ähnelt, ohne andere Differenzierung.3 Nach Murphey et al.,4 die histologischen Kriterien für die Diagnose eines Myxoms wurden 1948 von Stout festgelegt, der das Myxom als „ein wahres Neoplasma definierte, das aus einem Mangel an Sternzellen in einem losen myxoiden Stroma aus Retikulin und Kollagenfasern besteht“.
Dies sind sehr seltene Tumoren, deren Ätiologie unbekannt ist, sie haben keine Vorliebe für Rasse und kein klares erbliches Muster. Der Beginn liegt zwischen dem vierten und siebten Lebensjahrzehnt, im Allgemeinen bei Frauen.5 Dies stimmt mit den Merkmalen unserer Patienten überein.
Myxome, die aus dem Skelettmuskel entstehen, werden als intramuskuläre Myxome bezeichnet und wurden 1965 von Erzinger und Weiss beschrieben.6 Die häufigste Lokalisation sind die langen Muskeln der oberen Extremitäten (50%-60%).7 Ihr Erscheinen in den Gesäßmuskeln, wie in einem unserer Fälle, ist nicht ungewöhnlich und dies zusammen mit dem Becken, ist die zweithäufigste Position.
Die häufigste Darstellungsform ist eine einzelne Läsion, obwohl es Fälle von multiplen Myxomen gibt, die normalerweise mit anderen Entitäten assoziiert sind und interessante klinisch-pathologische Bilder bilden.
Mazabraud-Syndrom8 ist definiert als die Assoziation von multiplen intramuskulären Myxomen und fibröser Knochendysplasie, die mono- oder poly-ostotisch sein kann. In unserem zweiten Fall war dieses Syndrom die Hauptverdachtsdiagnose, obwohl wir keine Knochenläsionen zur endgültigen Bestätigung identifizierten. Es sollte nicht vergessen werden, dass sie später auftreten können, daher ist eine Röntgenüberwachung unerlässlich, da fibröse Knochendysplasien im Gegensatz zu Myxomen bösartig werden können. Das McCune-Albright9-Syndrom ist eine weitere zu berücksichtigende Störung. Dieser Zustand besteht aus multiplen Myxomen, polyostotischer fibröser Dysplasie, Café-au-Lait-Flecken und endokriner Überfunktion (vorzeitige Pubertät).
Hierbei handelt es sich um einen langsam wachsenden Tumor, der sich meist als schmerzlose, feste und bewegliche Masse darstellt. Wenn Schmerzen auftreten, wird dies durch Kompression der umgebenden Strukturen verursacht, wie in einigen Fällen in der Literatur beschrieben.7
Die für die Diagnose erforderlichen bildgebenden Tests sind einfaches Röntgen, CAT und MRT. Röntgenstrahlen sind in der Regel normal, CAT und MRT zeigen charakteristische Befunde, einschließlich der intramuskulären Lokalisation, fettähnlicher Gewebsgrenzen und hohem Wassergehalt. So zeigt es sich abgeschwächt auf CAT, hypointensiv in T1-gewichteten Bildern und hyperintensiv in T2-gewichteten Bildern.10
Vor der Operation wird eine histologische Diagnose mit Feinnadelaspiration oder Open-Sky-Biopsie empfohlen.11 Obwohl, wie wir im ersten Fall gesehen haben, dieser Test nicht endgültig ist.
Die Behandlung ist eine breite Resektion der Läsionen. Einige Autoren bevorzugen einen konservativeren Ansatz für kleine Massen, da dies gutartige Läsionen sind und eine Masse erst entfernen, wenn sie schmerzhaft oder groß wird. Bei dem ersten unserer Patienten haben wir uns aufgrund der Größe der Läsion für eine aggressivere Behandlung entschieden, während wir uns bei dem zweiten Patienten dafür entschieden haben, die Läsionen einzeln zu entfernen. Es gibt keine Artikel in der Literatur, die den Beginn von Metastasen oder Malignität beschreiben. Rezidivfälle sind auf Enukleation oder unvollständige Resektion zurückzuführen.12
Makroskopisch handelt es sich um weiche, eiförmige oder globuläre Massen, die von Kollagen und myxoidem Material abhängig sind, gallertartig, gelegentlich mit mit Flüssigkeit gefüllten Zystenräumen und von Skelettmuskelbündeln oder Fasziengewebe bedeckt sind.
Zytologisch zeichnen sie sich durch ihre myxoide Basis und geringe und gutartige Zellularität aus. Sie sollten jedoch von anderen Entitäten myxoiden Ursprungs unterschieden werden, entweder gutartig (noduläre Fasziitis, Neurotekom) oder bösartig (Lyposarkom, Fibrohistiozytom, myxoides Chondrosarkom) und von intramuskulären Metastasen muzinöser Adenokarzinome.
Schlussfolgerungen
Dies ist ein sehr seltener mesenchymaler Tumor unbekannter Ätiologie, der in den langen Muskeln der oberen Extremitäten oder des Beckens auftritt. Es ist normalerweise eine einzelne Läsion, aber das Vorhandensein mehrerer Läsionen könnte auf einen Teil eines Syndroms hindeuten. Die Behandlung ist eine Operation, es besteht kein Malignitätsrisiko und ein Wiederauftreten ist auf eine unvollständige Exzision der Läsion zurückzuführen. Die endgültige Diagnose ist anatomopathologisch.
Ethische Offenlegungenschutz von Menschen und Tieren
Die Autoren erklären, dass die angewandten Verfahren den Vorschriften der zuständigen Ethikkommission für klinische Forschung und denen des Ethikkodex der Weltärztekammer (Erklärung von Helsinki) entsprachen.
Vertraulichkeit der Daten
Die Autoren erklären, dass sie die Protokolle ihres Arbeitszentrums zur Veröffentlichung von Patientendaten befolgt haben.
Recht auf Privatsphäre und Einwilligung nach Aufklärung