Einleitung
Der kruppelähnliche Faktor 4 (KLF4) gehört zur Familie der Zinkfinger-Transkriptionsfaktoren, die in verschiedenen menschlichen Geweben exprimiert werden. Es ist bekannt als einer der vier Faktoren der Induktion zu pluripotenten Stammzellen (iPSCs) (Ghaleb und Yang, 2017). KLF4 kann mehrere wichtige biologische Prozesse wie Neuroinflammation, oxidativen Stress, Proliferation, Differenzierung und Apoptose regulieren (Kaushik et al., 2010; Mamonkin et al., 2013; Zhang et al., 2015; Miao et al., 2017; Xu et al., 2017). Viele frühere Studien konzentrierten sich auf die Rolle von KLF4 bei der Krebsentstehung und -progression (Karam et al., 2017; Yadav et al., 2018). KLF4 ist ein Transkriptionsfaktor mit zwei Funktionen, der je nach Krebsart oder Krebsstadium seine Rolle als Onkogen oder Tumorsuppressorgen ausüben kann (Evans und Liu, 2008). Es kann die Transkription von Genen aktivieren oder hemmen, die an der Zellproliferation, Differenzierung und Apoptose beteiligt sind (Ding et al., 2015). KLF4 kann mit anderen Umprogrammierungsfaktoren zusammenarbeiten, um die somatischen Zellen in iPSCs umzuwandeln und die Differenzierung von Stammzellen zu hemmen (Takahashi und Yamanaka, 2006; van Schaijik et al., 2018). Dies bietet therapeutische Perspektiven für Gefäßerkrankungen, Immunerkrankungen, Anorexie und andere Krankheiten (Imbernon et al., 2014; Liu Y. et al., 2015; Murgai et al., 2017). Darüber hinaus kann KLF4 eine wichtige regulatorische Rolle im zentralen Nervensystem (ZNS) spielen. Mehrere Studien weisen darauf hin, dass KLF4 mit mehreren neurologischen Störungen verbunden ist, einschließlich Alzheimer-Krankheit (AD), Epilepsie, Parkinson-Krankheit, Hydrozephalus und Schizophrenie (Qin et al., 2011; Xie et al., 2013; Han et al., 2015; Nishiguchi et al., 2015; Li L. et al., 2017).
AD ist eine der häufigsten chronischen neurodegenerativen Erkrankungen, die zu kognitiven und Gedächtnisstörungen, verschiedenen psychischen Symptomen und Verhaltensstörungen führt, und fortschreitende Demenz ist das häufigste klinische Merkmal (Jiang et al., 2018). Zu den derzeit bestätigten pathogenen Faktoren der AD gehören die Bildung seniler Plaques, die durch abnormale Amyloid-β (Aß) -Ablagerung induziert werden, und die durch Tau-Akkumulation induzierten neurofibrillären Verwicklungen oder dystrophischen Neuritiden (Querfurth und LaFerla, 2010; Shinohara et al., 2014). Darüber hinaus kann AD auch durch genetische Faktoren beeinflusst werden. Die Pathogenese ist jedoch noch unklar. Die am weitesten verbreiteten Medikamente zur Behandlung von AD sind Neurotransmitter-Enhancer, Anti-Amyloid-Mittel, neuroprotektive Peptide und andere Medikamente (Cacabelos, 2018). Bemerkenswerterweise haben mehrere Studien gezeigt, dass KLF4 eine bedeutende Rolle bei der Pathogenese von AD spielte. In dieser Übersicht konzentrieren wir uns auf die regulatorische Rolle von KLF4 bei Neuroinflammation, neuronaler Apoptose, axonaler Regeneration und Eisenakkumulation, um den Zusammenhang zwischen KLF4 und der Pathogenese von AD zu erklären, der Einblicke in die zellulären und molekularen Mechanismen neurodegenerativer Erkrankungen geben könnte.
Die biologischen Eigenschaften von KLF4
KLF4 ist ein Zinkfinger-haltiges Kernprotein, das aus der NIH 3T3-Bibliothek isoliert und im Zellkern lokalisiert ist. Es wurde zuerst von Shields et al. (1996). Die Molekülmasse von menschlichem KLF4 beträgt 55kD und befindet sich auf dem Chromosom 9q31. KLF4 deckt ein 6,3 kb großes Gensegment ab und hat fünf Exons. Seine cDNA-kodierende Region kodiert für ein Polypeptid, das aus 470 Aminosäureresten besteht (Yet et al., 1998; Ghaleb und Yang, 2017). Der Carboxyterminus von KLF4 weist eine DNA-Bindungsstrukturregion auf, die drei Zinkfingerstrukturen vom Typ Cys2His2 (C2H2) enthält, die von 81 hochkonservierten Aminosäuren gebildet werden. Es reguliert die Transkription durch hohe Affinität zu CACCC-Elementen und GC-reichen Zielgen-DNA-Sequenzen (Shields und Yang, 1998; Pearson et al., 2008). Die meisten DNA-Bindungsstellen von KLF4 befinden sich innerhalb der Zinkfingerregion, einschließlich der N-terminalen Transkriptionsaktivierungsdomäne für interagierende Proteine, der C-terminalen Zinkfingerstruktur für die DNA-Bindung und der Transkriptionshemmungszone (Bieker, 2001). KLF4 ist an der Regulierung der Expression vieler endogener Gene beteiligt (Shields und Yang, 1998). Am Aminoterminus von KLF4 befindet sich eine hochvariable Transkriptionsregulationsdomäne. Die Aminosäurereste, die sich zwischen der 91- und der 117-Aminosäure befinden, bilden eine transkriptionelle Aktivierungsdomäne, die reich an Prolin und Serin ist, während auch eine transkriptionelle Repressionsdomäne existiert. Daher hat KLF4 zwei nachteilige Wirkungen: Aktivierung und Hemmung der Gentranskription (Yet et al., 1998; Wei et al., 2006).
Während der Embryonalentwicklung wurde KLF4 im späten Stadium der Embryonalentwicklung höher exprimiert. In reifen Geweben und Organen wird KLF4 hauptsächlich im Magen-Darm-Trakt, in der Mundhöhle, in der Hautepidermis, im Gefäßendothel und in der Niere exprimiert und im Gehirn weniger exprimiert (Segre et al., 1999; Ghaleb et al., 2011; Liu et al., 2013; Chen et al., 2015; Er et al., 2015; Bin et al., 2016). Es wird angenommen, dass es eine wichtige Rolle bei der Regulierung der Zellproliferation und -differenzierung spielt. Außerdem kann KLF4 auch den Zellzyklus regulieren. KLF4 kann P21 in einer P53-abhängigen Weise aktivieren (Zhang et al., 2000). Darüber hinaus wurde festgestellt, dass KLF4 (–/–) –Zellen früher in die Seneszenzphase eintraten als KLF4 (+/+) –Zellen, was durch die geringere antioxidative Genexpression und den höheren Gehalt an reaktiven Sauerstoffspezies (ROS) in KLF4 (-/-) -Zellen erklärt werden kann. ROS kann die p53- und p21-Expression erhöhen und anschließend die DNA-Schädigung fördern (Liu C. et al., 2015). Es wurde festgestellt, dass PRMT5 die KLF4-Expression in den Proteinspiegeln erhöhen kann. Es wurde berichtet, dass PRMT5 die Transkription von p21 erhöht und die Expression von bax durch Hemmung der KLF4-Ubiquitylierung verringert (Hu et al., 2015). Darüber hinaus haben zahlreiche Studien gezeigt, dass KLF4 an der Regulation der Apoptose von Neuronen beteiligt ist (Kong et al., 2016; Kui et al., 2017; Song et al., 2018). Die regulatorische Rolle von KLF4, die wir kennen, ist noch wenig und weitere Untersuchungen sind erforderlich.
Rolle von KLF4 bei AD
Es ist allgemein bekannt, dass AD hauptsächlich durch Gedächtnis- und kognitive Beeinträchtigungen und exekutive Dysfunktion gekennzeichnet ist (Goedert und Spillantini, 2006). Viele Studien haben gezeigt, dass neuronale Apoptose und synaptische Dysfunktion sind pathologische Grundlage für den Rückgang der kognitiven Funktion (Caccamo et al., 2017; Guo et al., 2017; Yoon et al., 2018). Die akkumulierten Schäden durch Aß-Ablagerung, oxidativen Stress und Eisenansammlung können zu neuronalen Funktionsstörungen und Apoptose von AD-Patienten führen. Mehrere Studien haben gezeigt, dass die regulatorische Rolle von KLF4 im ZNS von entscheidender Bedeutung zu sein scheint. In Anbetracht der Tatsache, dass KLF4 neuronale Apoptose, synaptische Regeneration, oxidativen Stress und Neuroinflammation reguliert, könnte die Beziehung zwischen KLF4 und der Pathogenese von AD ein potenzielles neues Ziel für die AD-Behandlung sein.
Rolle von KLF4 bei der Neuroinflammation
Zahlreiche klinische Studien haben gezeigt, dass Aß aggregieren kann und der Hauptbestandteil der extrazellulären Ablagerungen des Hirngewebes von AD-Patienten ist, die die umgebenden Synapsen und Neuronen beeinträchtigen und zum neuronalen Tod führen können. Eine abnormale Sekretion oder übermäßige Produktion von Aß führt zu pathologischen Veränderungen der AD, daher ist die Aß-Ablagerung das Kernglied der AD (Rajmohan und Reddy, 2017). Darüber hinaus haben Studien gezeigt, dass eine übermäßige Aß-Ablagerung Gliazellen dazu anregen kann, ROS und andere Einflussfaktoren abzusondern, was zu oxidativem Stress führt. Es war bekannt, dass oxidativer Stress die Produktion von Aß stimulieren kann. Daher können Aß und oxidativer Stress miteinander interagieren und das Fortschreiten der AD beeinflussen (Chevignon et al., 2018).
KLF4 wurde als potenzieller Modulator berichtet und hat eine große Wirkung auf Entzündungen durch Vermittlung von Makrophagen und Endothelzellen (Abbildung 1) (Yoshida et al., 2014; Kapoor et al., 2015; Yang et al., 2018). Im ZNS können übermäßige und chronische Entzündungsreaktionen Schäden an Neuronen und Neurogliozyten verursachen. Es wurde kürzlich gezeigt, dass die KLF4-Expression positiv mit Aß42-induzierter Neuroinflammation korreliert. In mikroglialen BV2-Zellen kann oligomeres Aß42 die KLF4-Expression erhöhen, die durch aktiviertes P53 vermittelt wird (Li L. et al., 2017). Unter entzündlichen Bedingungen, wie der Aß-Akkumulation, kann die Freisetzung von proinflammatorischen Zytokinen bei der Erzeugung von AD stimuliert werden (Griffin und Barger, 2010). Die Neurotoxizitätspotenz und die entzündungsfördernde Potenz löslicher Aß42-Oligomere ist relativ höher als die der unlöslichen Ballaststoffe (Selkoe, 1991; Weinberg et al., 2018). Die Stille von KLF4 kann die Aß42-vermittelte Neuroinflammation wiederherstellen, und eine Überexpression von KLF4 kann die Aß42-vermittelte Neuroinflammation verschlimmern (Li L. et al., 2017). Die Aß-Akkumulation induziert die Aktivierung von Astrozyten und Mikroglia (Rodríguez et al., 2016). Aktivierte Astrozyten können die Neuroinflammation verstärken, indem sie entzündungsfördernde Faktoren wie IL-1, IL-6 und TNF-α freisetzen (Rubio-Perez und Morillas-Ruiz, 2012; Doméné et al., 2016). Der Teufelskreis der Entzündungsreaktionen führt schließlich zu Funktionsstörungen und neuronaler Apoptose.
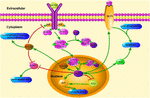
ABBILDUNG 1. Schematische Darstellung von KLF4-bezogenen Signalwegen. Diese Zahl unterstreicht die Rolle von KLF4 bei der Neuroprotektion und Axonregeneration. Die Pfeile in der Abbildung zeigen Aktivierung oder Förderung an, und die geraden Linien zeigen verwandte Hemmung an. KLF4, Kruppelähnlicher Faktor 4; STAT3, Signalwandler und Aktivator der Transkription 3; JAK, Januskinase; SOCS3, Suppressor der Zytokinsignalisierung 3; HCP1, Häm-Trägerprotein 1; ERK5, mitogenaktivierte Proteinkinase 5.
KLF4 spielt eine entscheidende Rolle bei der Regulierung entzündungsfördernder Signale. In Gliazellen erhöht die durch Gemfibrozil induzierte KLF4-Aktivierung den Suppressor des Zytokinsignals 3 (SOCS3) über den PI3-Kinase-AKT-Weg (Ghosh und Pahan, 2012). Der siRNA-vermittelte Knockdown von KLF4 könnte das Niveau von SOCS in Astroglia und Mikroglia von Mäusen abschwächen, was anschließend die Expression von Entzündungsgenen beeinflussen könnte (Kaushik et al., 2010; Ghosh und Pahan, 2012). Darüber hinaus kann die SOCS-Deletion das Überleben verletzter Neuronen fördern und die Axonregeneration fördern (Smith et al., 2009; Sun et al., 2011). Und KLF4 reguliert positiv die Produktion von IL-1β oder anderen entzündungsfördernden Markern. Es reguliert positiv die Cyclooxygenase-2 (Cox-2) und negativ die induzierbare Stickoxidsynthase (iNOS) (Kaushik et al., 2013). Darüber hinaus ist KLF4 ein wichtiger regulatorischer Faktor für die Monozytendifferenzierung und ein potenzielles Ziel für die Immunregulation (Alder et al., 2008). Daher könnte KLF4 die Neuroinflammation fördern, indem es diese negativen Regulatoren reguliert.
Es ist erwähnenswert, dass KLF4 im Parkinson-Krankheitsmodell MPP + -induzierten oxidativen Stress und Neurotoxizität fördern und dann die neuronale Apoptose erhöhen und die Zellproliferation verzögern kann (Chen et al., 2013). Oxidativer Stress ist ein Ungleichgewicht zwischen Peroxidation und Antioxidation. Freie Radikale können Veränderungen in verschiedenen Makromolekülen verursachen, die zu Zellschäden, Zellalterung und Gewebeschäden führen (Parajuli et al., 2013; Nie et al., 2015). Oxidativer Stress kann eine frühe Entzündung und Aß-Produktion verschlimmern und dann die AD verschlimmern (Cai et al., 2011). Daher kann KLF4 an oxidativem Stress bei AD beteiligt sein.
Diese Ergebnisse implizieren KLF4 eine Schlüsselrolle bei der Vermittlung von Neuroinflammation durch Aktivierung der Mikroglia und die damit verbundene Freisetzung von proinflammatorischen Zytokinen. Es hat das Potenzial, die Neuroinflammation zu verstärken. Bisher haben sich viele Studien zur Pathogenese von AD auf die Neuroinflammation konzentriert. Als potenzielles Ziel für die Immunregulation kann KLF4 die Entzündungsreaktionen von Mikroglia über die Beeinflussung verwandter negativer Regulatoren fördern, was einen großen Einfluss auf die Entwicklung von AD hat.
Rolle von KLF4 bei der Apoptose
Neurodegenerative Veränderungen umfassen den allmählichen Verlust von Neuronen und Synapsen in den repräsentativen Hirnregionen wie der Großhirnrinde, dem Hippocampus und anderen subkortikalen Regionen. Die durch neuronalen Verlust induzierten Funktionsstörungen des ZNS sind dauerhaft (Citron, 2010). Anhaltender oxidativer Stress kann zu neuronaler Apoptose führen (Wu et al., 2010). Eine große Anzahl von Studien hat bestätigt, dass AD eng mit oxidativem Stress zusammenhängt (Lee et al., 2012; Yui et al., 2015). Es wurde festgestellt, dass chronischer oxidativer Stress die Expression von Phospholipase A2 Gruppe 3 (Pla2g3) in Astrozyten verstärken und das Gleichgewicht von Aß stören und folglich zur Entwicklung von AD führen kann (Yui et al., 2015).
Viele Studien haben gezeigt, dass KLF4 eine wichtige Rolle bei der Hemmung der Entwicklung von oxidativem Stress spielt (Shi et al., 2014; Liu C. et al., 2015). Es wurde gefunden, dass KLF4 die durch H2O2 induzierte Zellapoptose fördern kann, diese Wirkung wird wahrscheinlich durch erhöhte Bax-Expression und verminderte bcl-2-Expression verursacht (Li et al., 2010). Quercetin könnte die KLF4-Expression in menschlichen Neuroblastom-SH-SY5Y-Zellen reduzieren und die Expression des bcl-2 / Bax-Verhältnisses erhöhen. Darüber hinaus kann Quercetin die Apoptoserate der SH-5YSY-Zelle moderieren und die Caspase-3-Enzymaktivität reduzieren (Xi et al., 2012). Eine aktuelle Studie untersuchte die neuroprotektive Wirkung der Mitogen-aktivierten Protein (MAP) Kinase 5 (ERK5) gegen oxidativen Stress. Die Aktivierung von ERK5 kann den Tod von H2O2-induzierten Hippocampusneuronen teilweise reduzieren und die NGF- und PC-induzierte Neuroprotektion erhöhen (Su et al., 2014). Nils et al. verwendete eine Mutante von MEK5 (MEK5D), um die ERK5-aktivierten Transkriptions- und Funktionsantworten in menschlichen Endothelzellen zu untersuchen, und identifizierte KLF4 als neuartiges Downstream-ERK5-Ziel (Ohnesorge et al., 2010). Es wurde festgestellt, dass eine Überexpression von KLF4 TNF-vermittelte Entzündungsreaktionen unterdrücken und die Leukozytenadhäsion und die Basalzellapoptose reduzieren kann. Diese Ergebnisse bestätigen, dass KLF4 entzündungshemmende und antiapoptotische Eigenschaften besitzt (Ohnesorge et al., 2010). Nachfolgende Experimente haben gezeigt, dass das Verschwinden der zerebralen kavernösen Malformation 1 (CCM1) in Endothelzellen ERK5 über den MEKK3-MEK5-Signalweg aktiviert und die KLF4-Expression erhöht (Cuttano et al., 2016). ERK5 spielt eine vermittelnde Rolle bei der Vorkonditionierung (PC) und dem Nervenwachstumsfaktor (NGF), der die Expression von KLF4 hochreguliert (Su et al., 2014). Darüber hinaus kann der RNAi-vermittelte Abbau von KLF4 auch die NGF- oder PC-induzierte Neuroprotektion reduzieren. Eine Überexpression von KLF4 führt zu einem höheren bcl-2/Bax-Verhältnis in H2O2-gestressten Zellen (Su et al., 2014). Überexprimiertes KLF4 beschleunigt Veränderungen in bcl-2 und bax durch Kombination mit seinem entsprechenden Promotor (Li et al., 2010). Die ERK5 / KLF4-Kaskade kann als Dreh- und Angelpunkt in verschiedenen Signalwegen fungieren, die Neuronen vor dem durch oxidativen Stress verursachten Tod schützen (Su et al., 2014).
Oxidativer Stress wurde als eng mit vielen degenerativen Erkrankungen verbunden angesehen. KLF4 spielt eine wichtige Rolle bei der Aufrechterhaltung der genomischen Stabilität bei oxidativem Stress. KLF4 und ERK5 wirken zusammen, um Neuronen vor oxidativem Stress-induzierter Apoptose zu schützen. Daher kann KLF4 als therapeutisches Ziel gegen oxidativen Stress wirken, wenn es aktiviert wird. Es wurde berichtet, dass Statin-Medikamente ERK5 aktivieren können, was zur Expression von KLF4 und seinen abhängigen Genen führt (Ohnesorge et al., 2010), aber der Mechanismus bleibt unklar, und KLF4-bezogene Upstream- und Downstream-Zielgene werden bei oxidativem Stress weniger untersucht, es besteht Bedarf an weiteren Studien.
Rolle von KLF4 bei der Axonregeneration
Früher Axonverlust ist ein häufiges Merkmal neurodegenerativer Erkrankungen. Synaptischer Verlust und Transportstörungen bei AD können kognitive Beeinträchtigungen verursachen (Holtzman et al., 2011; Coleman, 2013). Der Grad der deklarativen Gedächtnisschädigung hängt mit der synaptischen Dichte im Hippocampus und im Kortex zusammen. Lösliche Aß-Oligomere reduzieren die Glutamataufnahme und fördern die synaptische Dysfunktion, wodurch die synaptische Plastizität gestört wird (Li et al., 2009). Daher ist es besonders wichtig zu untersuchen, wie die Axone im ZNS repariert werden können. In retinalen Ganglienzellen haben Axone eine starke Fähigkeit, während der frühen Entwicklung zu wachsen und sich zu regenerieren, aber im ZNS erwachsener Säugetiere verlieren Axone ihre Regenerationskapazität und die Neuronen können absterben oder verkümmern (Goldberg und Barres, 2000; Goldberg et al., 2002).
KLF4 spielt eine wichtige Rolle bei der Hemmung des Axonwachstums. In embryonalen RGCs kann eine Überexpression von KLF4 den Prozentsatz der Neuritenverlängerung, die Länge von Axonen und Dendriten und die Neuritenverzweigung verringern. Außerdem wurde festgestellt, dass die Überexpression von KLF4 die langfristigen postnatalen Axonwachstumsraten reduzieren kann, aber nicht die kurzfristigen Axonwachstumsraten (Moore et al., 2009; Steketee et al., 2014). Spätere Studien haben ergeben, dass die Axonbündel von KLF4–cKO-Mäusen dicker waren als Kontrollmäuse (Fang et al., 2016). Darüber hinaus kann die Entfernung der KLF4-Expression während der Entwicklung das Reproduktionspotenzial von adulten RGCs erhöhen. Darüber hinaus hatte KLF4 ohne die c-terminale DNA-Bindungsdomäne keinen Einfluss auf das Axonwachstum. Es gab keinen Einfluss auf das Überleben von Zellen, nachdem retinale Ganglienzellen verletzt wurden, wenn das KLF4 ausgeknockt wurde (Moore et al., 2009).
KLF4 kann auch die axonale Regeneration beeinflussen. Eine kürzlich durchgeführte Studie berichtete, dass die Abnahme der KLF4-Expression in adulten retinalen Ganglienzellen die Axonregeneration über den JAK-STAT3-Signalweg förderte (Qin et al., 2013). KLF4 erhöhte die Phosphorylierung von STAT3 und regulierte das Axonwachstum über JAK-STAT-Signalisierung (Qin und Zhang, 2012). Unter der Behandlung von Zytokinen werden Mitglieder der STAT-Proteinfamilie an den carboxyterminalen Tyrosin- und Serinstellen innerhalb der Zelle phosphoryliert, um ein stabiles Dimer zu bilden. Diese Modifikation verstärkt die Transkription von Zell-assoziierten Genen (Yuan et al., 2005). Die Wechselwirkung zwischen KLF4 und STAT3 bei der zytokininduzierten Phosphorylierung von Tyrosin705 hemmt die Expression von STAT3 durch Hemmung der Bindung von STAT3 an DNA (Qin et al., 2013). KLF4 Knockdown verbessert offensichtlich die Regeneration des Axons in den Ganglienzellen der Netzhaut nach einer Verletzung des Sehnervs und verhindert, dass der Nerv nach einer leichten Hirnverletzung verletzt wird. Die Aktionen werden durch eine Abnahme von p-p53 und eine Erhöhung der pSTAT3-Spiegel vermittelt. KLF4 reguliert die neuronale Apoptose positiv über die p53- und JAK-STAT3-Pfade und KLF4 reguliert die axonale Reparatur negativ über den JAK-STAT3-Pfad (Cui et al., 2017).
Daher stellten wir die Hypothese auf, dass bei AD die axonale Regeneration durch Veränderung der Expression von KLF4 oder durch Veränderung der intrazellulären Signalwege und durch Kontrolle der AD-Progression durch Verringerung fehlender Axone oder Verringerung der axonalen Dysfunktion erreicht werden kann. Die Verwendung des KLF4-Transkriptionsfaktors in potenziellen Therapeutika bedarf jedoch noch weiterer Untersuchungen.
Rolle von KLF4 bei der Eisenakkumulation
Eisen ist in biologischen Systemen weit verbreitet, Eisen-verwandte Metalloproteinasen spielen eine Schlüsselrolle beim Transport von Sauerstoff, beim Transfer von Elektronen und beim Katalysieren biochemischer Reaktionen (Aisen et al., 2001). Ein Überschuss an Eisen über den normalen physiologischen Bereich hinaus kann jedoch die menschliche Gesundheit schädigen (Adlard und Bush, 2006). Studien haben ergeben, dass der Eisengehalt im Hippocampus negativ mit der Leistung von Gedächtnistests korreliert (Ding et al., 2009). Eine erhöhte Eisenlast im Gehirn beschleunigt die Bildung von Aß-Plaques und hyperphosphorylierten Tau-Verwicklungen und verstärkt gleichzeitig den oxidativen Stress (Peters et al., 2015). Eisen, das ein hohes Maß an Permeabilität aufweist, fördert das Nervenwachstum und die Zell-Zell-Verbindungen während der Gehirnentwicklung (Dallman und Spirito, 1977).
Eine kürzlich durchgeführte Studie zeigte, dass physiologischer Stress die Aktivierung des KLF4-HCP1-Signalwegs und eine erhöhte Häm-Aufnahme verursachte (Li H. et al., 2017). Häm macht 95% des funktionellen Eisens im menschlichen Körper aus. Es ist eine der Hauptkomponenten der Hämoxygenase (Hooda et al., 2014; Kurucz et al., 2018). Eine Erhöhung der Aktivität von Oxygenase-1 kann die Oxidation des alternden Gehirns verzögern (Verdile et al., 2015; Serini und Calviello, 2016; Kurucz et al., 2018). Dies hat eine entlastende Wirkung auf AD. Physiologischer Stress induziert einen Anstieg des Glucocorticoidspiegels, Glucocorticoid erhöht die Expression von Häm-Trägerprotein 1 (HCP1) über KLF4, und dann fördert HCP1 die Häm-Aufnahme (Li H. et al., 2017). Glukokortikoid und KLF4 regulieren gemeinsam entzündungshemmende Gene, und Zellen mit niedrigem Glukokortikoidgehalt können die KLF4-Expression nicht vollständig induzieren (Sevilla et al., 2015). KLF4-induzierte Erhöhung der Häm-Aufnahme führt zu Eisenansammlung im Gehirn. Eisen fördert die Freisetzung von ROS (Tronel et al., 2013). Eisenelement erhöht Gehirn oxidativen Stress bei Ratten unter psychischem Stress (Yu et al., 2011). Daher kann HCP1 durch KLF4 und Glucocorticoid zusammen reguliert werden. Eine Erhöhung von HCP1 erhöht die Häm-Aufnahme, was direkt zur Eisenansammlung im Gehirn führt, die Oxidation verschlimmert, die Apoptose oder Dysfunktion erhöht und die Hirnschädigung verschlimmert.
Es ist allgemein anerkannt, dass die Gedächtnis- und Lernbehinderung sind die Hauptsymptome von AD. Eine große Anzahl klinischer Daten hat gezeigt, dass Aß-Plaquebelastung und Eisenakkumulationsreaktion auf die Entwicklung von Lern- und kognitiven Dysfunktionen bei AD (van Bergen et al., 2018). Kürzlich veröffentlichte Daten deuten darauf hin, dass hochdosiertes Eisen die Aß-Ablagerung erhöht und das Lernen und Gedächtnis bei Mäusen abschwächt (Guo et al., 2013). Klinische Studien haben gezeigt, dass eisenhaltige Mikroglia im Hippocampus von AD-Patienten unter Magnetresonanztomographie gefunden werden (Zeineh et al., 2015). Mikroglia erwerben Eisen aus übertragenden oder nicht übertragenden, extrazellulären und intrazellulären Quellen (McCarthy et al., 2018). Selektive und anhaltende KLF4-Expression kann im Zellkern und Zytoplasma ischämischer hippocampaler reaktiver Astrozyten induziert werden (Park et al., 2014). Studien haben gezeigt, dass KLF4 als transkriptioneller Repressor wirkt. Es reguliert die Expression von ELK-3 herunter und dann hemmt ELK-3 die Expression von HO-1 (Tsoyi et al., 2015). Hämoxygenase-1 (HO-1) ist ein Stressprotein, das Häm zu Bilirubin, freiem Eisen und Kohlenmonoxid abbaut. Die Hochregulation von HO-1 in Astrozyten kann zu abnormaler Eisenablagerung und mitochondrialer Dysfunktion im Gehirn führen, was zu einer verminderten kognitiven Fähigkeit führt (Schipper, 1999, 2004). Daher kann KLF4 am Prozess der Eisenakkumulation in Astrozyten beteiligt sein, was die Oxidation bei AD verschlimmert und Hirnschäden verschlimmert.
Schlussfolgerung
Es ist allgemein bekannt, dass KLF4 eine entscheidende Rolle bei der Regulierung der Zellproliferation, Apoptose und Differenzierung spielt. Frühere Studien konzentrierten sich auf die Regulation von KLF4 in mehreren wichtigen neurophysiologischen Prozessen, einschließlich Neuroinflammation, Neuroprotektion und synaptischer Regeneration. Kürzlich wurde festgestellt, dass KLF4 eine wichtige Rolle bei der Pathogenese von AD spielt. In diesem Artikel, Wir überprüfen die Rolle von KLF4 bei der Neuroprotektion und Neurogenese bei AD.
KLF4 ist nicht nur ein Regulator der Regulation der Zellproliferation und -differenzierung, sondern auch ein potenzielles Ziel für die Regulierung der Immunantwort. KLF4 kann negative Entzündungsfaktoren regulieren und Entzündungsreaktionen fördern und einen großen Einfluss auf die Expression von Astrozyten-Kernmikroglia haben. Darüber hinaus können KLF4 und ERK5 zusammen wirken, um neuroprotektive Wirkungen auszuüben. Darüber hinaus kann die Axonregeneration durch Veränderung des Gehalts spezifischer Transkriptionsfaktoren, intrazellulärer Inhibitoren oder durch Veränderung intrazellulärer Signalwege erreicht werden. Das Ausschalten von KLF4 kann die Axonregeneration verbessern und die Axonwachstumsrate beschleunigen. Die Reduktion der KLF4-Expression fördert die Axonregeneration über den JAK-STAT3-Weg, und KLF4 fördert den JAK-STAT3-Weg zur weiteren Axonregeneration. Daher könnte KLF4 am Prozess der entzündungshemmenden, Anti-Apoptose, Axonregeneration und Eisenakkumulation im ZNS beteiligt sein, der eine entscheidende Rolle bei der AD-Erzeugung spielt. Diese Ergebnisse legen nahe, dass KLF4 ein potenzielles therapeutisches Ziel für AD darstellt. Die tiefen zellulären und molekularen Mechanismen der Auswirkungen von KLF4 auf AD bleiben jedoch unklar und weitere Untersuchungen sind erforderlich.
Autorenbeiträge
ZQC, XHZ und YJ schrieben das Manuskript. SHG und JYL modifizierten den Rahmen des Manuskripts. BJL und RJC lieferten die kritischen Revisionen. Alle Autoren genehmigten die endgültige Version des Manuskripts zur Einreichung.
Finanzierung
Diese Arbeit wurde durch Zuschüsse der Natural Science Foundation of China (NSFC) (81871070, 31571126, 31471120) und der Jilin Science and Technology Agency Funding (Grant No. 20180519003JH, 20180414051GH, 20170414034GH und 20180414050GH).
Interessenkonflikterklärung
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Cacabelos, R. (2018). Gab es Verbesserungen in der Alzheimer-Krankheit Drug Discovery in den letzten 5 Jahren. Verwendbar bis. Opin. Droge Discov. 13, 523–538. doi: 10.1080/17460441.2018.1457645
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Liu, Y., Shi, J. und Chen, X. (2015). Identifizierung neuer Targets für saisonale allergische Rhinitis während und außerhalb der Pollensaison durch Microarray-Analyse. Acta Otolaryngol. 135, 1330–1336. doi: 10.3109/00016489.2015.1067906
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Serini, S. und Calviello, G. (2016). Reduktion von oxidativem / nitrosativem Stress im Gehirn und seine Beteiligung an der neuroprotektiven Wirkung von n-3 PUFA bei Alzheimer. Curr. Alzheimer Res. 13, 123-134. doi: 10.2174/1567205012666150921101147
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar