Einführung
Das L1-Zelladhäsionsmolekülgen (L1CAM) ist ein neuronales Zelladhäsionsmolekül, das zur Immunglobulin-Superfamilie gehört. es besitzt Schlüsselfunktionen bei der Entwicklung des Nervensystems (Itoh und Fushiki, 2015). Mutationen in L1CAM wurden mit X-verknüpften neurologischen Syndromen in Verbindung gebracht, die als L1-Krankheiten zusammengefasst werden. Sie werden wie folgt klassifiziert: X-verknüpfter Hydrozephalus (XLH) aufgrund einer Stenose des Aquädukts von Sylvius (HSAS), MASA-Syndrom (geistige Behinderung, Aphasie, schlurfender Gang, adduzierte Daumen), spastische Paraparese Typ 1 (SP1) und X-verknüpfte Agenesie des Corpus callosum (ACC) (Weller und Gartner, 2001; Itoh und Fushiki, 2015).
Über 282 krankheitsverursachende Mutationen (DMs) im L1CAM-Gen wurde in HGMD® Professional 2019.2 berichtet (https://portal.biobase-international.com/hgmd/pro/all.php). Veränderungen im L1CAM-Gen sind vielfältig; Mutationsdatenanalysen von 282 Patienten zeigen 51% Missense- und Nonsense-Mutationen, 25% Deletionen, 5% Insertionen und 19% Spleißstellenänderungen, aber stille Mutationen in L1CAM mit pathogenem Potenzial waren selten, und stille Mutationen wurden häufig ignoriert, insbesondere beim Nachweis der Whole-Exome-Sequenzierung (WES).
In dieser Studie untersuchten wir mit WES die fetale DNA einer chinesischen schwangeren Frau, die fünf kontinuierliche Schwangerschaften mit fetalem Hydrozephalus gemeldet hat; Wir fanden nur eine neuartige stille Mutation c.453G > T (p.Gly151 = ) im L1CAM-Gen. Interessanterweise zeigten wir durch weitere Analysen, dass die stille Mutation eine potentielle 5′-Spleißstellen-Konsensussequenz erzeugte, die zu einer In-Frame-Deletion von 72 bp aus Exon 5 und 24 Aminosäuren des L1CAM-Proteins führen würde.
Fallvorstellung
Eine 28-jährige gesunde Frau wurde nach vier freiwilligen Schwangerschaftsabbrüchen aufgrund eines fetalen Hydrozephalus in anderen Krankenhäusern an unsere Klinik überwiesen. Alle Föten waren männlich. Als sie in unserem Krankenhaus (Frauenkrankenhaus, Medizinische Fakultät, Zhejiang University, Zhejiang, China) ankam, befand sie sich bereits in der fünften Schwangerschaftswoche mit einem fetalen Hydrozephalus durch Bilduntersuchungen. Um die genetische Ursache zu untersuchen, wurde eine fetale Blutentnahme im Gestationsalter von 26 Wochen durchgeführt. Konventionelle zytogenetische Studien wurden sowohl für fetale als auch für elterliche Proben durchgeführt, und die fetale Probe wurde weiter analysiert durch Einzelnukleotidpolymorphismus (SNP) Array und WES.
Diese Studie wurde in Übereinstimmung mit den Empfehlungen der Ethikkommission des Frauenkrankenhauses der Medizinischen Fakultät der Zhejiang-Universität durchgeführt, und von allen Teilnehmern dieser Studie wurde eine Einverständniserklärung gemäß der Erklärung von Helsinki eingeholt. Das Studienprotokoll wurde vom Prüfungsausschuss des Frauenkrankenhauses der Medizinischen Fakultät der Zhejiang-Universität in China genehmigt.
Materialien und Methoden
Karyotyp und SNP-Array
Die Karyotypen von fetalem Nabelschnurblut und peripherem Nabelschnurblut wurden durch konventionelle Karyotypisierung von mindestens 30 Blutlymphozyten bestimmt, die an der Metaphase durch Colchicine angehalten wurden. G-Banding-Karyotypen von kultivierten Zellen wurden auf der 320-400-Band-Ebene mit einer Auflösung von etwa 10 Mb durchgeführt. Das SNP-Array wurde vom CytoScan ™ HD-Array (Affymetrix, USA) gemäß den Anweisungen des Herstellers mit rund 2.600.000 Markern einschließlich 750.000 SNP-Sonden und 1.900.000 Nicht-Polymorphismus-Sonden für eine umfassende Abdeckung des gesamten Genoms durchgeführt. Die Daten wurden mit der Chromosome Analysis Suite (ChAS) -Software (Affymetrix, Santa Clara, CA) basierend auf der GRCh37 / hg19-Assembly analysiert. Die Meldeschwelle für das Ergebnis der Kopienzahl wurde auf 500 kb mit einer Markerzahl ≥50 für Gewinne und auf 200 kb mit einer Markerzahl ≥50 für Verluste festgelegt.
Whole-Exome Sequencing
Der Hauptteil von WES wurde vom Beijing Genomics Institute bereitgestellt. Genomische DNA wurde mit einem DNeasy Blood Kit (Qiagen, CA) extrahiert und dann mit Covaris LE220 (Massachusetts, USA) fragmentiert, um eine gepaarte Endbibliothek (200-250 bp) zu erzeugen. Alle amplifizierten Bibliotheken wurden auf der BGISEQ-500-Plattform durchgeführt, die Einzelstrang-DNA wurde mit dem MGIEasy ™ DNA Library Prep Kit V1 (BGI, Shenzhen, China) gemischt und anschließend unter Verwendung der 100SR-Chemie mit dem BGISEQ-500RS High-Throughput Sequencing Kit (BGI, Shenzhen, China) sequenziert.
Saubere Lesevorgänge (mit einer Länge von 90 bp), die aus gezielter Sequenzierung und Filterung abgeleitet wurden, wurden dann mit dem Burrows-Wheeler Aligner (BWA) Multi-Vision-Softwarepaket (Li und Durbin, 2009) auf die Referenz des menschlichen Genoms (hg19) ausgerichtet. Nach der Ausrichtung wurden die Ausgabedateien verwendet, um eine Sequenzierungsabdeckung und eine Tiefenanalyse der Zielregion, der Einzelnukleotidvarianten (SNVs) und der Indels durchzuführen, und wir verwendeten die GATK-Software, um SNVs und Indels zu erkennen (McKenna et al., 2010), alle SNVs und Indels wurden gefiltert und geschätzt über mehrere Datenbanken, einschließlich der National Center for Biotechnology Information (NCBI) Single-Nucleotide Polymorphism Database (dbSNP), HapMap, 1000 Genome Project Dataset und Datenbank von 100 chinesischen gesunden Erwachsenen. Wir verwendeten Condel, SIFT, Polyphenol-2, LRT, Mutation Taster und PhyloP, um die Wirkung von Varianten vorherzusagen. Pathogene Varianten werden nach dem vom American College of Medical Genetics and Genomics (ACMG) herausgegebenen Protokoll bewertet (Richards et al., 2015). Die Human Gene Mutation Database (HGMD) wurde verwendet, um Mutationen zu screenen. Alle potenziellen pathogenen Varianten wurden mit Sanger-Sequenzierungsmethoden validiert.
RNA-Extraktion, PCR und Sequenzierung
Periphere mononukleäre Blutzellen (PMBCs) und mononukleäre Nabelschnurblutzellen (CBMCs) wurden durch Ficoll-Dichtegradiententrennung isoliert. Die Gesamt-RNA wurde aus PMBCs und den CBMCs mit TRIzol (Takara, Japan) extrahiert. Extrahierte Gesamt-RNAs wurden mittels RT-Kit (Takara, Japan) reverse transkribiert. Die PCR wurde mit GoldStar Best Master Mix (CWBIO, Beijing) durchgeführt. Primersequenzen sind aufgelistet: L1CAM-DNA-5F, CCCACCCGTCCTTTCCTA; L1CAM-DNA-5R, CGCTCGTCCTGCTTGATGT; L1CAM-mRNA-4-6- F, GGTGTCCACTTCAAACCCAA; und L1CAM-mRNA-4-6- R, GCGGCTTCCTGTCAATCA. Die Sanger-Sequenzierung wurde mit einem ABI 3130 DNA-Analysator durchgeführt.
Ergebnisse
Eine 28-jährige gesunde Frau wurde nach vier freiwilligen Schwangerschaftsabbrüchen aufgrund eines fetalen Hydrozephalus an unsere Klinik überwiesen. Alle Feten waren männlich (Abbildung 1A). Der familiäre Stammbaum schien XLH zu zeigen. Sie war bereits in der fünften Schwangerschaft bei 26 Schwangerschaftswochen. Fetale Ventrikulomegalie wurde durch fetale Ultraschalluntersuchung und MRT nachgewiesen, was durchweg das Vorhandensein von Hydrozephalus zeigte. Sie zeigten, dass der bilaterale Hirnventrikel und der dritte Ventrikel offensichtlich erweitert waren und ein schwerer Hydrozephalus in der intrazerebralen und Agenese des Corpus callosum auftrat (Abbildung 1B).
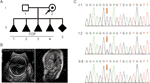
Abbildung 1 (A) Stammbaum der Familie. TOP, Schwangerschaftsabbruch. (B) Bildgebende Untersuchungen des Fötus. Fetale Ultraschalluntersuchung und fetale MRT zeigten einen schweren Hydrozephalus im Fötus. (C) Sequenzanalyse von genomischer DNA von Familienmitgliedern. Die Genotypen von L1CAM waren Wildtyp, c.453G > T Het, und c.453G > T Hom, in I:1 (Ehemann), I: 2 (schwangere Frau) und II: 5 (Fötus). Die Mutation wird durch die roten Pfeile angezeigt.
Um die mögliche genetische Ursache zu untersuchen, führten wir eine Karyotypanalyse und ein SNP-Array durch, um die fetale Blutentnahme zu analysieren, und fanden keine positiven Ergebnisse. Das Ergebnis der choroidalen Neovaskularisation (CNV) wurde im Gene Expression Omnibus (GEO) hinterlegt; Die Zugangsnummer lautet GSE133063, wie unten angehängt (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
Dann entdeckten wir den Fötus durch WES. Die analytische Strategie zum Auffinden einer wahrscheinlichen Identifizierung pathogener Varianten wurde in Abbildung S1 gezeigt. Eine Liste von Varianten (Tabelle S1) wurde durch das Screening von Variantenhäufigkeiten, Mutationsstatus und Vererbungsmodus erhalten. Unter Berücksichtigung der Hydrocephalus-assoziierten Gene (HP:0000238, http://compbio.charite.de/hpoweb/showterm?id=HP:0000238#id=HP_0000238) (Tabelle S2) gab es außer der stillen Mutation von c.453G > T im Exon 5 des L1CAM-Gens (NM_000425.3) keine weitere bemerkenswerte Mutation. c.453G > T wurde in HGMD und ClinVar nicht gemeldet und in dbSNP, gnomAD und anderen Datensätzen nicht gefunden. Nach den Standards und Richtlinien der ACMG (Richards et al., 2015) hatte es das Kriterium „pathogen“ oder „wahrscheinlich pathogen“ noch nicht erreicht, aber es gab keine anderen potenziellen Mutationen; Wir hatten keine andere Wahl, als eine weitere Analyse der gefundenen stillen Mutation durchzuführen.
Nach traditionellem Denken trat diese Basenersetzung in der dritten Base im Codon 151 auf, die ein Glycin kodiert, wodurch eine neutrale Mutation erzeugt wurde (p.Gly151 = ). Diese Variante wurde in DNA bestätigt, die aus fötalem Nabelschnurblut und peripherem Blut des Paares durch Sanger-Sequenzierung extrahiert wurde (Abbildung 1C). Die Frau trug die heterozygote Mutation, und ihr Ehemann war ein Wildtyp-Genotyp.
Mit Mutation Taster (http://www.mutationtaster.org/) wurde c.453G > T als „krankheitsverursachend“ bewertet.“ Es zeigte sich, dass Proteinmerkmale betroffen sein könnten und die Spleißstelle verändert werden könnte; Wir waren neugierig auf die möglichen Spleißeffekte der L1CAM-Funktion in dieser stillen Mutation. Die stille Mutation wurde mit den folgenden Online-Softwareprodukten getestet: NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) und NNSplice (http://www.fruitfly.org/seq_tools/splice.html); Es wurde auch vorhergesagt, dass die potenzielle 5′-Spleißstelle in der L1CAM c erstellt wird.453G > T Mutation unter Verwendung der Softwareprodukte (Abbildung S2). Die Ergebnisse zeigten, dass diese stille Mutation eine potentielle 5′-Spleißstelle 72 bp stromaufwärts von der normalen Exon 6 / Intron 6-Spleißstelle erzeugte (Abbildung 2A). Wenn dies der Fall ist, können wir die Längenänderung von L1CAM Messenger RNA (mRNA) zwischen I: 2 und II: 5 finden (Abbildung 2A). Die RT-PCR wurde unter Verwendung von Primern durchgeführt, die zur Amplifikation der Exons 4-6 in der L1CAM-mRNA bestimmt waren. Tatsächlich zeigten die Ergebnisse eine kurze Verkürzungsbande in der fetalen cDNA-PCR (II: 5), während die aus der L1CAM-mRNA amplifizierte Bande die erwartete lange Bande in der fetalen cDNA-PCR (I) enthielt:1) und lange/kurze Banden in der cDNA-PCR der Schwangeren (I:2) (Abbildung 2B). Die direkte Sequenzierung des amplifizierten Fragments zeigte, dass die Deletion die letzten 72 bp von Exon 5 in männlicher fetaler cDNA beinhaltete (die Frau war ein Träger) (Abbildung 2C). Wir haben die entscheidenden pathogenen Beweise.
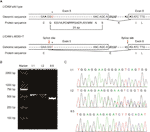
Abbildung 2 (A) Schematische Darstellung von Exon 5, Intron 6 und Exon 6-Organisation in L1CAM. (B) RT-PCR-Analyse der Exons 5 und 6 der L1CAM-cDNA aus peripheren mononukleären Blutzellen (PMBCs) und mononukleären Nabelschnurblutzellen (CBMCs). Agarosegelelektrophorese von RT-PCR-Produkten, die aus I: 1 (Ehemann), I: 2 (schwangere Frau) und II: 5 (Fötus) erzeugt wurden. (C) Sequenzanalyse des RT-PCR-Produkts aus PMBCs des Paares und CBMCs des Fötus.
Diese stille Mutation führte zu 24 Aminosäuren des L1CAM-Proteins (Reste 151-174); Lys (K) wurde am Codon 175 durch Glu (E) ersetzt (Abbildung 2A). Es gab die Ausrichtung mehrerer L1CAM-Proteinsequenzen über mehrere Arten hinweg und die Erhaltung der fehlenden Aminosäuren in L1CAM bei Säugetieren: Homo sapiens, Pan troglodytes, Bos taurus, Mus musculus und Rattus norvegicus (Abbildung 3A). Wildtyp- und c.453G > T-Spleißmutation L1CAM-Proteine wurden mit der Software CPHmodels-3.2 Server (http://www.cbs.dtu.dk/services/CPHmodels/) vorhergesagt (Abbildung 3B). Immunglobulinähnliche (Ig-ähnliche) Domäne 2 (Reste 134-230) von Wildtyp- und Spleißmutations-L1CAM-Proteinen ist in Abbildung 3C dargestellt. L1CAM c.453G > Die Spleißmutation veränderte die Proteinstruktur, insbesondere die Ig-ähnliche Domäne 2

Abbildung 3 (A) Ausrichtung mehrerer L1CAM-Proteinsequenzen über Spezies hinweg. Die L1CAM c.453G > T führte dazu, dass 24 Aminosäuren des L1CAM-Proteins (Reste 151-174) in der konservierten Aminosäureregion in verschiedenen Spezies fehlten. Die schwarze Spalte zeigt die fehlenden Aminosäuren. (B) Die Strukturen des Wildtyp- und c.453G > T-Spleißmutations-L1CAM-Proteins, wie von der Software CPHmodels-3.2 Server vorhergesagt. (C) Die Strukturen der immunglobulin-ähnlichen (Ig-ähnlichen) Domäne 2 (Reste 134-230) des Wildtyp- und Spleißmutations-L1CAM-Proteins.
>
Stille Mutationen wurden häufig von WES entdeckt, aber es wurde nicht genügend Aufmerksamkeit geschenkt, was zum Weglassen von DMs führte. In dieser Studie beschäftigten wir WES, um die genetische Ursache einer chinesischen Familie mit Hydrocephalus zu untersuchen, fanden aber nur eine neuartige stille Mutation in L1CAM, die uns zu einer weiteren Analyse zwang. Glücklicherweise haben wir bewiesen, dass die stille Mutation eine neue 5′-Spleißstelle schuf und ein DM war.
Mutationen in L1CAM können eine X-chromosomale L1-Krankheit verursachen, aber die klinischen Symptome sind variabel; Mutationen erzeugen unerwartete Phänotypen. In der Studie sind die fünf leidenden Föten alle Männer, was mit einem Vererbungsmuster übereinstimmt. Der fetale Ultraschall und die MRT zeigen eine typische L1-Erkrankung, einschließlich XLH und Agenese des Corpus callosum. Es verbessert unser Verständnis der Genotyp-Phänotyp-Korrelation von L1CAM.
L1CAM c.453G > T (p.Gly151 = ) wurde zunächst angenommen, dass es keinen Einfluss auf die Proteinsequenz hat. Es wurde jedoch berichtet, dass andere stille Mutationen, c.924C > T (p.Gly308 = ) und c.645C > T (p.Gly215 = ), im L1CAM-Gen DMs sind (Du et al., 1998; Vos et al., 2010). Das c.924C > T-Mutation führte zur Aktivierung einer neuen Spleißstelle 69 bp 5′ zur normalen Spleißstelle des Exon 8 / Intron 8-Spenders und wurde als „krankheitsverursachende“ Stelle für Hydrocephalus deklariert (Du et al., 1998). Für c.645C > T in L1CAM wurden 51 bp mit der Aktivierung eines neuen Exon 6/Intron 6 Donor Splice deletiert (Vos et al., 2010). Unsere vorliegende Studie war ähnlich; Die Mutation von c.453G > T erzeugte eine potentielle 5′-Spleißstelle stromaufwärts der normalen Exon 5 / Intron 5-Spleißstelle. Alle diese stillen Mutationen erzeugten neue Spenderspleißstellen, was dazu führte, dass das Exon übersprungen wurde. Es erinnerte uns daran, genau auf diese stillen Mutationen zu achten, die das Spleißen von Proteinen beeinflussen können.
Als Transmembran-Glykoprotein und Mitglied der Immunglobulin-Superfamilie der Zelladhäsionsmoleküle kann das L1CAM-Protein an der Zelloberfläche mit einer Reihe verschiedener Glykoproteine interagieren, und homophile Bindung ist wahrscheinlich seine Hauptart der Interaktion (Wei und Ryu, 2012). Die Untersuchungen zur Kristallstruktur der Ig-ähnlichen Domänen 1-4 in Neurofascin legen nahe, dass viele pathologische L1-Mutationen konservierte Aminosäurereste innerhalb dieser Domänen beeinflussen und homophile Wechselwirkungen stören (Liu et al., 2011), insbesondere wie durch die Funktionsforschung der Ig-ähnlichen Domäne 2 (Zhao et al., 1998). In unserer Studie spekulierten wir, dass L1CAM c.453G > T die Ig-ähnliche Domäne 2 im extrazellulären Teil des L1CAM-Proteins veränderte, was zu einer abnormalen extrazellulären Interaktion führte und nicht in der Lage war, stromabwärts den Signalweg zu initiieren. Eine weitere Studie, die sich der Massenspektrometrie dieser L1CAM-Variante widmet, würde genau klären, welches molekulare Ensemble in der Zelle produziert wird.
Zusammenfassend berichteten wir über WES über eine neuartige stille Mutation c.453G > T in L1CAM, die eine 5′-Spleißstelle erzeugt, die für den Hydrozephalus verantwortlich ist. Es wurde vorhergesagt, dass diese abnormale Proteinvariante die Ig-ähnliche Domäne 2 verändert, was die homophile Bindung des L1CAM-Proteins beeinflussen könnte. Darüber hinaus führten wir eine pränatale genetische Diagnose für die schwangere Frau durch, die fünf kontinuierliche Schwangerschaften mit Hydrozephalus meldete. In der Zwischenzeit schlug es vor, dass einige stille Mutationen, die in WES entdeckt wurden, nicht ignoriert werden sollten; Spleißvorhersagen dieser Mutationen waren notwendig. Es bot eine neue genetische Grundlage für die Pränataldiagnostik und die Pränataldiagnostik des Hydrozephalus vor der Implantation.
Datenverfügbarkeit
Öffentlich verfügbare Datensätze wurden in dieser Studie analysiert. Diese Daten finden Sie hier: GSE133063(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
Ethikerklärung
Die Studien mit menschlichen Teilnehmern wurden vom Prüfungsausschuss des Frauenkrankenhauses der Medizinischen Fakultät der Zhejiang-Universität in China überprüft und genehmigt. Die schriftliche Einverständniserklärung zur Teilnahme an dieser Studie wurde vom gesetzlichen Vormund / den nächsten Angehörigen der Teilnehmer abgegeben. Die schriftliche Einverständniserklärung der Person (en) und des gesetzlichen Vormunds / der nächsten Angehörigen der Minderjährigen wurde für die Veröffentlichung potenziell identifizierbarer Bilder oder Daten in diesem Artikel eingeholt.
Autorenbeiträge
YS, YLi, MC, YLu, YQ und YY führten Experimente durch. YS hat die Zahlen vorbereitet. MC und YLi analysierten die WES-Daten. YLu und YQ führten eine Karyotypanalyse und ein SNP-Array durch. YY rekrutierte Proben. HL und FL lieferten bildgebende Untersuchungen. YS und MD schrieben das Manuskript. Alle Autoren haben das endgültige Manuskript gelesen und genehmigt.
Finanzierung
Diese Studie wurde von der National Natural Science Foundation of China (Grant No. 81801441), dem wichtigsten Forschungs- und Entwicklungsprogramm der Provinz Zhejiang (Grant No. 2019C03025), das nationale Forschungs- und Entwicklungsprogramm Chinas (Grant No. 2016YFC1000703) und die Medical Scientific Research Foundation der Provinz Zhejiang (Grant No. 2014KYA246).
Interessenkonflikterklärung
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Danksagung
Wir danken den Patienten, die an dieser Studie teilgenommen haben. Wir danken Dr. Jiong Gao (BGI Genomics, BGI-Shenzhen, Shenzhen 518083, China) für seine Unterstützung bei der Erstellung dieses Manuskripts.
Ergänzungsmaterial
Das Ergänzungsmaterial zu diesem Artikel finden Sie online unter: https://www.frontiersin.org/articles/10.3389/fgene.2019.00817/full#supplementary-material
Abbildung S1 / Analytische Strategie zum Auffinden einer wahrscheinlichen Identifizierung pathogener Varianten durch WES.
Abbildung S2 / Spenderspleißstellen, die von NetGene2 und NNSplice vorhergesagt wurden.
Tabelle S1 / Liste der Varianten durch das Screening von Variantenhäufigkeiten, Mutationsstatus und Vererbungsmodus.
Tabelle S2 / Liste der Hydrocephalus-assoziierten Gene (Export für HP:0000238).
Du, YZ, Dickerson, C., Aylsworth, AS, Schwartz, CE (1998). Eine stille Mutation, C924T (G308G), im L1CAM-Gen führt zu X-Linked Hydrocephalus (HSAS). Dr. Med. Genet. 35 (6), 456–462. Ursprungsbezeichnung: 10.1136/jmg.35.6.456
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Itoh, K., Fushiki, S. (2015). Die Rolle von L1cam in der murinen Kortikogenese und die Pathogenese des Hydrocephalus. Pathologisch. Int. 65 (2), 58–66. doi: 10.1111/Stift.12245
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Li, H., Durbin, R. (2009). Schnelle und genaue kurze Leseausrichtung mit Burrows-Wheeler Transform. Bioinformatik 25 (14), 1754-1760. doi: 10.1093/bioinformatik/btp324
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Liu, H., Focia, PJ, Er, X. (2011). Homophiler Adhäsionsmechanismus von Neurofascin, einem Mitglied der L1-Familie von neuronalen Zelladhäsionsmolekülen. In: J. Biol. Chem. 286 (1), 797–805. Ursprungsbezeichnung: 10.1074/jbc.M110.180281
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitationen von McKenna, A., Hanna, M., Sivachenko, A., Kernytsky, A., et al. (2010). Das Genomanalyse-Toolkit: Ein MapReduce-Framework zur Analyse von DNA-Sequenzierungsdaten der nächsten Generation. Genome Res. 20 (9), 1297-1303. Ursprungsbezeichnung: 10.1101/gr.107524.110
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitation Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards und Richtlinien für die Interpretation von Sequenzvarianten: eine gemeinsame Konsensempfehlung des American College of Medical Genetics and Genomics und der Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi: 10.1038/gim.2015.30
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Vos, Y. J., de Walle, H. E. K., Bos, K. K., Stegeman, J. A., Berge, A. M., Bruining, M., et al. (2010). Genotyp-Phänotyp-Korrelationen beim L1-Syndrom: ein Leitfaden für genetische Beratung und Mutationsanalyse. Dr.Med. Genet. 47 (3), 169–175. doi: 10.1136/jmg.2009.071688
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Wei, C. H., Ryu, S. E. (2012). Homophile Wechselwirkung der L1-Familie von Zelladhäsionsmolekülen. Verwendbar bis. Mol. Med. 44 (7), 413–423. Ursprungsbezeichnung: 10.3858/emm.2012.44.7.050
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Weller, S., Gartner, J. (2001). Genetische und klinische Aspekte des X-chromosomalen Hydrozephalus (L1-Krankheit): Mutationen im L1CAM-Gen. Summen. Mutanten. 18 (1), 1–12. doi: 10.1002/humu.1144
PubMed Abstract | CrossRef Full Text | Google Scholar
Zhao, X., Yip, P. M., Siu, C. H. (1998). Identification of a homophilic binding site in immunoglobulin-like domain 2 of the cell adhesion molecule L1. J. Neurochem. 71 (3), 960–971 doi: 10.1046/j.1471-4159.1998.71030960.x.
PubMed Abstract | CrossRef Full Text | Google Scholar