- Introducción
- Las características biológicas de KLF4
- Papel de KLF4 en la EA
- Papel de KLF4 en la neuroinflamación
- El papel de KLF4 en la apoptosis
- El papel de KLF4 en la regeneración de axones
- Papel de KLF4 en la acumulación de hierro
- Conclusión
- Contribuciones de los autores
- Financiación
- Declaración de Conflicto de Intereses
Introducción
El factor 4 similar a Kruppel (KLF4) es un miembro de la familia del factor de transcripción de dedos de zinc,que se expresa en varios tejidos humanos. Es bien conocido como uno de los cuatro factores de la inducción a células madre pluripotentes (IPSC) (Ghaleb y Yang, 2017). KLF4 puede regular múltiples procesos biológicos importantes, como neuroinflamación, estrés oxidativo, proliferación, diferenciación y apoptosis (Kaushik et al., 2010; Mamonkin et al., 2013; Zhang et al., 2015; Miao et al., 2017; Xu et al., 2017). Cantidades de estudios previos se centraron en el papel de KLF4 en el desarrollo y la progresión del cáncer (Karam et al., 2017; Yadav et al., 2018). KLF4 es un factor de transcripción de doble función, que puede ejercer su papel como un oncogén o un gen supresor de tumores dependiendo del tipo de cáncer o estadio del cáncer (Evans y Liu, 2008). Puede activar o inhibir la transcripción de genes implicados en la proliferación celular, la diferenciación y la apoptosis (Ding et al., 2015). KLF4 puede colaborar con otros factores de reprogramación para convertir las células somáticas en IPSC e inhibir la diferenciación de células madre (Takahashi y Yamanaka, 2006; van Schaijik et al., 2018). Esto proporciona perspectivas terapéuticas para enfermedades vasculares, enfermedades inmunitarias, anorexia y otras enfermedades(Imbernon et al., 2014; Liu Y. et al., 2015; Murgai et al., 2017). Además, KLF4 puede desempeñar un papel ampliamente regulador en el sistema nervioso central (SNC). Varios estudios indican que KLF4 está relacionado con múltiples trastornos neurológicos, incluyendo la enfermedad de Alzheimer( EA), epilepsia, enfermedad de Parkinson, hidrocefalia y esquizofrenia (Qin et al., 2011; Xie et al., 2013; Han et al., 2015; Nishiguchi et al., 2015; Li L. et al., 2017).
La EA es una de las enfermedades neurodegenerativas crónicas más comunes, que conduce a deficiencias cognitivas y de memoria, varios síntomas mentales y anormalidades conductuales, y la demencia progresiva es la característica clínica más común (Jiang et al., 2018). Los factores patógenos confirmados actuales de EA incluyen la formación de placas seniles inducidas por deposición anormal de β-amiloide (Aß) y los enredos neurofibrilares o neuritis distrófica inducida por acumulación de tau (Querfurth y LaFerla, 2010; Shinohara et al., 2014). Además, la EA también puede verse afectada por factores genéticos. Sin embargo, la patogénesis inducida sigue siendo oscura. Los medicamentos más prevalentes para el tratamiento de la EA incluyen potenciadores de neurotransmisores, agentes antiamiloides, péptidos neuroprotectores y otros medicamentos (Cacabelos, 2018). En particular, varios estudios han demostrado que KLF4 desempeñó un papel importante en la patogénesis de la EA. En esta revisión, nos centramos en el papel regulador de KLF4 en la neuroinflamación, la apoptosis neuronal, la regeneración axonal y la acumulación de hierro para explicar la asociación entre KLF4 y la patogénesis de la EA, lo que podría proporcionar información sobre los mecanismos celulares y moleculares de los trastornos neurodegenerativos.
Las características biológicas de KLF4
KLF4 es una proteína nuclear que contiene dedos de zinc, aislada de la biblioteca NIH 3T3 y ubicada en el núcleo celular. Primero fue identificado y caracterizado por Shields et al. (1996). La masa molecular del KLF4 humano es de 55 KD y se encuentra en el cromosoma 9q31. KLF4 cubre un segmento genético de 6,3 kb y tiene cinco exones. Su región codificante del ADNc codifica un polipéptido que consta de 470 residuos de aminoácidos (Yet et al., 1998; Ghaleb y Yang, 2017). El término carboxi de KLF4 tiene una región de estructura de unión al ADN que contiene tres estructuras de dedo de zinc de tipo Cys2His2 (C2H2), que están formadas por 81 aminoácidos altamente conservados. Regula la transcripción por alta afinidad con elementos CACCC y secuencias de ADN de genes diana ricos en GC (Shields y Yang, 1998; Pearson et al., 2008). La mayoría de los sitios de unión al ADN de KLF4 se encuentran dentro de la región del dedo de zinc, incluido el dominio de activación de transcripción N-terminal para las proteínas que interactúan, la estructura del dedo de zinc C-terminal para la unión al ADN y la zona de inhibición de la transcripción (Bieker, 2001). KLF4 participa en la regulación de la expresión de muchos genes endógenos (Shields y Yang, 1998). Hay un dominio regulador transcripcional altamente variable en el terminal amino de KLF4. Los residuos de aminoácidos ubicados entre el amino 91 y el amino 117 constituyen un dominio de activación transcripcional, que es rico en prolina y serina, mientras que también existe un dominio de represión transcripcional. Por lo tanto, KLF4 tiene dos efectos adversos: activar e inhibir la transcripción de genes (Yet et al., 1998; Wei et al., 2006).
Durante el desarrollo embrionario, KLF4 se expresó más alto en la etapa final del desarrollo embrionario. Mientras que en tejidos y órganos maduros, KLF4 se expresa principalmente en el tracto gastrointestinal, la cavidad oral, la epidermis de la piel, el endotelio vascular y el riñón, y se expresa menos en el cerebro (Segre et al., 1999; Ghaleb et al., 2011; Liu et al., 2013; Chen et al., 2015; He et al., 2015; Bin et al., 2016). Se cree que desempeña un papel importante en la regulación de la proliferación y diferenciación celular. Además, KLF4 también puede regular el ciclo celular. KLF4 puede activar P21 de una manera dependiente de P53 (Zhang et al., 2000). Además, se encontró que las células KLF4 (–/–) entraron en la fase de senescencia antes que las células KLF4 ( + / + ), lo que puede explicarse por la menor expresión génica antioxidante y el mayor nivel de especies reactivas de oxígeno (ROS) en las células KLF4 ( – / – ). ROS puede aumentar la expresión de p53 y p21 y posteriormente promover el daño al ADN(Liu C. et al., 2015). Se encontró que PRMT5 puede elevar la expresión de KLF4 en los niveles de proteínas. Se informó que la PRMT5 aumentaba la transcripción de p21 y disminuía la expresión de bax a través de la inhibición de la ubiquitilación de KLF4(Hu et al., 2015). Además, numerosos estudios han demostrado que KLF4 está involucrado en la regulación de la apoptosis de las neuronas (Kong et al., 2016; Cui et al., 2017; Song et al., 2018). El papel regulador fisiológico de KLF4 que hemos conocido es todavía poco y se necesitan más investigaciones.
Papel de KLF4 en la EA
Está bien establecido que la EA se caracteriza principalmente por deficiencias de memoria y cognitivas y disfunción ejecutiva (Goedert y Spillantini, 2006). Muchos estudios han demostrado que la apoptosis neuronal y la disfunción sináptica son la base patológica del deterioro de la función cognitiva (Caccamo et al., 2017; Guo et al., 2017; Yoon et al., 2018). El daño acumulado de la deposición de Aß, el estrés oxidativo y la acumulación de hierro pueden provocar disfunción neuronal y apoptosis en pacientes con EA. Varios estudios han demostrado que la función reguladora de KLF4 parece ser crucial en el SNC. Teniendo en cuenta que se informó que KLF4 regulaba la apoptosis neuronal, la regeneración sináptica, el estrés oxidativo y la neuroinflamación, la relación entre KLF4 y la patogénesis de la EA podría ser un nuevo objetivo potencial para el tratamiento de la EA.
Papel de KLF4 en la neuroinflamación
Cantidades de estudios clínicos han demostrado que el Aß puede agregarse y es el componente principal de los depósitos extracelulares del tejido cerebral de los pacientes con EA, lo que puede dañar las sinapsis y neuronas circundantes y provocar la muerte neuronal. La secreción anormal o la producción excesiva de Aß conduce a cambios patológicos de la EA, por lo que la deposición de Aß es el eslabón central de la EA (Rajmohan y Reddy, 2017). Además, los estudios han demostrado que la deposición excesiva de Aß puede estimular a las células gliales a secretar ROS y otros factores que influyen, lo que conduce al estrés oxidativo. Se sabía que el estrés oxidativo puede estimular la producción de Aß. Por lo tanto, el Aß y el estrés oxidativo pueden interactuar entre sí y afectar la progresión de la EA (Cheignon et al., 2018).
KLF4 fue reportado como un modulador potencial y tiene un gran efecto sobre la inflamación al mediar macrófagos y células endoteliales (Figura 1) (Yoshida et al., 2014; Kapoor et al., 2015; Yang et al., 2018). En el SNC, las reacciones inflamatorias excesivas y crónicas pueden causar daños en las neuronas y los neurogliocitos. Recientemente se demostró que la expresión de KLF4 se correlacionaba positivamente con la neuroinflamación inducida por Aß42. En las células microgliales BV2, el Aß42 oligomérico puede aumentar la expresión de KLF4, que está mediada por P53 activado (Li L. et al., 2017). En condiciones inflamatorias, como la acumulación de Aß, la liberación de citoquinas proinflamatorias puede ser estimulada en la generación de EA (Griffin y Barger, 2010). La potencia de neurotoxicidad y la potencia proinflamatoria de los oligómeros solubles Aß42 es relativamente más alta que el depósito de fibra insoluble (Selkoe, 1991; Weinberg et al., 2018). El silencio de KLF4 es capaz de restaurar la neuroinflamación mediada por Aß42, y la sobreexpresión de KLF4 puede exacerbar la neuroinflamación mediada por Aß42 (Li L. et al., 2017). La acumulación de Aß induce la activación de astrocitos y microglía (Rodríguez et al., 2016). Los astrocitos activados pueden potenciar la neuroinflamación al liberar factores proinflamatorios como IL-1, IL-6 y TNF-α (Rubio-Perez y Morillas-Ruiz, 2012; Doméné et al., 2016). El círculo vicioso de las respuestas inflamatorias eventualmente conduce a la disfunción y la apoptosis neuronal.
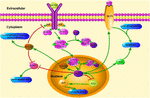
FIGURA 1. Ilustración esquemática de vías de señalización relacionadas con KLF4. Esta figura destaca el papel de KLF4 en la neuroprotección y la regeneración de axones. Las flechas de la figura indican activación o promoción, y las líneas rectas indican inhibición relacionada. KLF4, Factor 4 similar a Kruppel; STAT3, Transductor de señal y activador de transcripción 3; JAK, Quinasa Janus; SOCS3, Supresor de señalización de citoquinas 3; HCP1, proteína transportadora de hemo 1; ERK5, proteína quinasa activada por mitógenos 5.
KLF4 juega un papel crucial en la regulación de las señales proinflamatorias. En las células gliales, la activación KLF4 inducida por gemfibrozilo aumenta el supresor de la señal 3 de citoquinas (SOCS3) a través de la vía PI3-quinasa-AKT (Ghosh y Pahan, 2012). El derribo de KLF4 mediado por siRNA podría atenuar el nivel de SOC en astroglia y microglia de ratones, lo que podría afectar posteriormente la expresión del gen inflamatorio (Kaushik et al., 2010; Ghosh y Pahan, 2012). Además, la eliminación de SOC puede promover la supervivencia de neuronas lesionadas y promover la regeneración de axones (Smith et al., 2009; Sun et al., 2011). Y KLF4 regula positivamente la producción de IL-1β u otros marcadores proinflamatorios. Regula positivamente la ciclooxigenasa – 2 (Cox-2) y regula negativamente la óxido nítrico sintasa inducible (iNOS) (Kaushik et al., 2013). Además, KLF4 es un factor regulador importante para la diferenciación de monocitos y un objetivo potencial para la regulación inmune (Alder et al., 2008). Por lo tanto, KLF4 podría promover la neuroinflamación regulando estos reguladores negativos.
Vale la pena mencionar que en el modelo de enfermedad de Parkinson, KLF4 puede promover el estrés oxidativo y la neurotoxicidad inducidos por MPP+, y luego aumentar la apoptosis neuronal y retrasar la proliferación celular (Chen et al., 2013). El estrés oxidativo es un desequilibrio entre la peroxidación y la antioxidación. Los radicales libres pueden causar cambios en diferentes macromoléculas, lo que lleva a daño celular, envejecimiento celular y daño tisular (Parajuli et al., 2013; Nie et al., 2015). El estrés oxidativo puede agravar la inflamación temprana y la producción de Aß y luego agravar la EA(Cai et al., 2011). Por lo tanto, KLF4 puede estar implicado en el estrés oxidativo en la EA.
Estos hallazgos implican que KLF4 desempeña un papel clave en la mediación de la neuroinflamación mediante la activación de la microglía y la consiguiente liberación de citoquinas proinflamatorias. Tiene potencial para mejorar la neuroinflamación. Hasta ahora, muchos estudios sobre la patogénesis de la EA se han centrado en la neuroinflamación. Como objetivo potencial para la regulación inmune, KLF4 puede promover las respuestas inflamatorias de la microglía a través de reguladores negativos relacionados que afectan, lo que tiene un gran efecto en el desarrollo de la EA.
El papel de KLF4 en la apoptosis
Los cambios neurodegenerativos incluyen la pérdida gradual de neuronas y sinapsis en las regiones representativas del cerebro, como la corteza cerebral, el hipocampo y otras regiones subcorticales. Las deficiencias funcionales del SNC inducidas por la pérdida neuronal son permanentes (Citron, 2010). El estrés oxidativo sostenido puede llevar a la apoptosis neuronal (Wu et al., 2010). Un gran número de estudios han confirmado que la EA está estrechamente relacionada con el estrés oxidativo (Lee et al., 2012; Yui et al., 2015). Se encontró que el estrés oxidativo crónico puede mejorar la expresión de la fosfolipasa A2 grupo 3 (Pla2g3) en astrocitos y alterar el equilibrio de Aß, y en consecuencia conducir al desarrollo de AD (Yui et al., 2015).
Muchos estudios han demostrado que KLF4 juega un papel importante en la inhibición del desarrollo de estrés oxidativo (Shi et al., 2014; Liu C. et al., 2015). Se encontró que KLF4 puede promover la apoptosis celular inducida por H2O2, esta acción es probablemente causada por el aumento de la expresión de bax y la disminución de la expresión de bcl-2 (Li et al., 2010). La quercetina podría reducir la expresión de KLF4 en las células SH-SY5Y de neuroblastoma humano y aumentar la expresión de la relación bcl-2/bax. Además, la quercetina puede moderar la tasa de apoptosis de las células SH-5YSY y reducir la actividad de la enzima caspasa-3 (Xi et al., 2012). Un estudio reciente investigó el efecto neuroprotector de la quinasa 5 de la proteína activada por mitógenos (PAM) (ERK5) contra el estrés oxidativo. La activación de ERK5 puede reducir parcialmente la muerte de las neuronas hipocampales inducidas por H2O2 y aumentar la neuroprotección inducida por NGF y PC(Su et al., 2014). Nils et al. se utilizó un mutante de MEK5 (MEK5D) para estudiar la transcripción activada por ERK5 y las respuestas funcionales en células endoteliales humanas, y se identificó que KLF4 era un nuevo objetivo de ERK5 aguas abajo (Ohnesorge et al., 2010). Se encontró que la sobreexpresión de KLF4 puede suprimir las respuestas inflamatorias mediadas por TNF y reducir la adhesión leucocitaria y la apoptosis de células basales. Estos resultados confirman que KLF4 tiene propiedades antiinflamatorias y antiapoptóticas (Ohnesorge et al., 2010). Experimentos posteriores han demostrado que la desaparición de la malformación cavernosa cerebral 1 (CCM1) en las células endoteliales activa ERK5 a través de la vía de señal MEKK3-MEK5 y aumenta la expresión de KLF4 (Cuttano et al., 2016). ERK5 desempeña un papel mediador en el preacondicionamiento (PC) y el factor de crecimiento nervioso (NGF), que regula la expresión de KLF4 (Su et al., 2014). Además, el derribo de KLF4 mediado por ARNi también puede reducir la neuroprotección inducida por NGF o PC. La sobreexpresión de KLF4 conduce a una mayor relación bcl-2 / bax en células estresadas por H2O2 (Su et al., 2014). El KLF4 sobreexpresado acelera los cambios en bcl-2 y bax al combinarse con su promotor correspondiente (Li et al., 2010). La cascada ERK5 / KLF4 puede actuar como pivote en varias vías que protegen a las neuronas de la muerte inducida por estrés oxidativo (Su et al., 2014).
Se ha considerado que el estrés oxidativo está estrechamente relacionado con muchas enfermedades degenerativas. KLF4 juega un papel importante en el mantenimiento de la estabilidad genómica en el estrés oxidativo. KLF4 y ERK5 actúan juntos para proteger a las neuronas de la apoptosis inducida por estrés oxidativo. Por lo tanto, KLF4 puede actuar como diana terapéutica para actuar contra el estrés oxidativo cuando se activa. Se ha informado que los medicamentos con estatinas pueden activar ERK5, lo que conduce a la expresión de KLF4 y sus genes dependientes (Ohnesorge et al., 2010), pero el mecanismo sigue sin estar claro, y los genes diana relacionados con KLF4 aguas arriba y aguas abajo se estudian menos en estrés oxidativo, por lo que es necesario realizar más estudios.
El papel de KLF4 en la regeneración de axones
La pérdida temprana de axones es una característica común de las enfermedades neurodegenerativas. La pérdida sináptica y el deterioro del transporte en la EA pueden causar deterioro cognitivo (Holtzman et al., 2011; Coleman, 2013). El grado de daño de la memoria declarativa está relacionado con la densidad sináptica en el hipocampo y la corteza. Los oligómeros solubles de Aß reducen la absorción de glutamato y promueven la disfunción sináptica, alterando la plasticidad sináptica(Li et al., 2009). Por lo tanto, es particularmente importante estudiar cómo reparar los axones en el SNC. En las células ganglionares de la retina, los axones tienen una fuerte capacidad de crecer y regenerarse durante el desarrollo temprano, pero en el SNC de los mamíferos adultos, los axones pierden su capacidad de regeneración y las neuronas pueden graduarse para morir o atrofiarse (Goldberg y Barres, 2000; Goldberg et al., 2002).
KLF4 juega un papel importante en la inhibición del crecimiento de axones. En los RGC embrionarios, la sobreexpresión de KLF4 puede reducir el porcentaje de elongación de neuritas, la longitud de axones y dendritas, y la ramificación de neuritas. Además, se encontró que la sobreexpresión de KLF4 puede reducir las tasas de crecimiento de axones postnatales a largo plazo, pero no pudo reducir las tasas de crecimiento de axones a corto plazo (Moore et al., 2009; Steketee et al., 2014). Estudios posteriores han encontrado que los haces de axones de ratones KLF4–cKO eran más gruesos que los ratones de control (Fang et al., 2016). Además, la eliminación de la expresión de KLF4 durante el desarrollo puede aumentar el potencial reproductivo de los GRG adultos. Además, KLF4 carente del dominio de unión al ADN c-terminal no tuvo ningún efecto en el crecimiento del axón. No hubo impacto en la supervivencia de las células después de que las células ganglionares de la retina se lesionaran si el KLF4 estaba noqueando (Moore et al., 2009).
KLF4 también puede afectar a la regeneración axonal. Un estudio reciente informó que la disminución de la expresión de KLF4 en células ganglionares de la retina adultas promovía la regeneración de axones a través de la vía JAK-STAT3 (Qin et al., 2013). KLF4 aumentó la fosforilación de STAT3 y reguló el crecimiento de axones a través de la señalización JAK-STAT (Qin y Zhang, 2012). Bajo el tratamiento de citoquinas, los miembros de la familia de proteínas STAT son fosforilados en los sitios de tirosina y serina carboxi-terminales dentro de la célula para formar un dímero estable. Esta modificación mejora la transcripción de genes asociados a células (Yuan et al., 2005). La interacción entre KLF4 y STAT3 en la fosforilación inducida por citocinas de la tirosina 705 inhibe la expresión de STAT3 al inhibir la unión de STAT3 al ADN (Qin et al., 2013). KLF4 knockdown obviamente mejora la regeneración del axón en las células ganglionares de la retina después de una lesión del nervio óptico, y evita que el nervio se lesione después de una lesión cerebral leve. Las acciones están mediadas por una disminución de p-p53 y un aumento de los niveles de pSTAT3. KLF4 regula positivamente la apoptosis neuronal a través de las vías p53 y JAK-STAT3, y KLF4 regula negativamente la reparación axonal a través de la vía JAK-STAT3 (Cui et al., 2017).
Por lo tanto, planteamos la hipótesis de que en la EA, la regeneración axonal se puede lograr alterando la expresión de KLF4 o alterando las vías de señalización intracelulares relacionadas, y controlando la progresión de la EA mediante la reducción de los axones faltantes o la reducción de la disfunción axonal. Sin embargo, la forma de utilizar el factor de transcripción KLF4 en terapias potenciales aún necesita más exploración.
Papel de KLF4 en la acumulación de hierro
El hierro se encuentra ampliamente en sistemas biológicos, las metaloproteinasas relacionadas con el hierro desempeñan un papel clave en el transporte de oxígeno, la transferencia de electrones y la catalización de reacciones bioquímicas (Aisen et al., 2001). Sin embargo, cualquier exceso de hierro más allá del rango fisiológico normal puede dañar la salud humana (Adlard y Bush, 2006). Los estudios han encontrado que el contenido de hierro en el hipocampo está correlacionado negativamente con el rendimiento de las pruebas de memoria (Ding et al., 2009). El aumento de la carga de hierro en el cerebro acelera la formación de placas de Aß y enredos tau hiperfosforilados, al tiempo que aumenta el estrés oxidativo (Peters et al., 2015). El hierro, que tiene un alto grado de permeabilidad, promueve el crecimiento nervioso y las conexiones de célula a célula durante el desarrollo cerebral (Dallman y Spirito, 1977).
Un estudio reciente demostró que el estrés fisiológico causaba la activación de la vía de señalización KLF4-HCP1 y un aumento de la captación de hemo (Li H. et al., 2017). El hemo representa el 95% del hierro funcional en el cuerpo humano. Es uno de los principales componentes de la hemo oxigenasa (Hooda et al., 2014; Kurucz et al., 2018). El aumento de la actividad de la oxigenasa-1 puede retrasar la oxidación del cerebro envejecido (Verdile et al., 2015; Serini y Calviello, 2016; Kurucz et al., 2018). Esto tiene un efecto de alivio en el anuncio. El estrés fisiológico induce el aumento del nivel de glucocorticoides, el glucocorticoide aumenta la expresión de la proteína transportadora de hemo 1 (HCP1) a través de KLF4, y luego el HCP1 promueve la captación de hemo (Li H. et al., 2017). Los glucocorticoides y KLF4 regulan conjuntamente los genes antiinflamatorios, y las células con bajo contenido de glucocorticoides no pueden inducir por completo la expresión de KLF4 (Sevilla et al., 2015). El aumento de la ingesta de hemo inducido por KLF4 conduce a la acumulación de hierro en el cerebro. El hierro promueve la liberación de ROS (Tronel et al., 2013). El elemento de hierro aumenta el estrés oxidativo cerebral en ratas sometidas a estrés psicológico (Yu et al., 2011). Por lo tanto, el HCP1 puede ser regulado por KLF4 y glucocorticoide juntos. El aumento de la HCP1 aumenta la captación del hemo, lo que conduce directamente a la acumulación de hierro en el cerebro, exacerba la oxidación, aumenta la apoptosis o disfunción y empeora el daño cerebral.
Generalmente se acepta que la discapacidad de la memoria y el aprendizaje son los principales síntomas de la EA. Un gran número de datos clínicos han demostrado que la carga de placa de Aß y la respuesta de acumulación de hierro al desarrollo del aprendizaje y la disfunción cognitiva en la EA (van Bergen et al., 2018). Datos publicados recientemente han sugerido que, altas dosis de hierro aumentan la deposición de Aß y atenúan el aprendizaje y la memoria en ratones (Guo et al., 2013). Los estudios clínicos han demostrado que la microglía que contiene hierro se encuentra en el hipocampo de pacientes con EA bajo imágenes por resonancia magnética (Zeineh et al., 2015). La microglía adquiere hierro de fuentes extracelulares e intracelulares transferibles o no transferibles (McCarthy et al., 2018). La expresión selectiva y sostenida de KLF4 se puede inducir en el núcleo y el citoplasma de los astrocitos reactivos isquémicos del hipocampo (Park et al., 2014). Los estudios han demostrado que KLF4 actúa como represor transcripcional. Regula la expresión de ELK-3, y luego ELK-3 inhibe la expresión de HO-1 (Tsoyi et al., 2015). La hemo oxigenasa-1 (HO-1) es una proteína de estrés que degrada el hemo en bilirrubina, hierro libre y monóxido de carbono. La regulación ascendente de HO-1 en los astrocitos puede provocar depósitos anormales de hierro y disfunción mitocondrial en el cerebro, lo que lleva a una disminución de la capacidad cognitiva (Schipper, 1999, 2004). Por lo tanto, KLF4 puede estar involucrado en el proceso de acumulación de hierro en astrocitos, exacerbando la oxidación en la EA y agravando el daño cerebral.
Conclusión
Se sabe que KLF4 desempeña un papel fundamental en la regulación de la proliferación celular, la apoptosis y la diferenciación. Estudios previos se han centrado en la regulación de KLF4 en varios procesos neurofisiológicos importantes, incluyendo neuroinflamación, neuroprotección y regeneración sináptica. Recientemente, se ha descubierto que KLF4 desempeña un papel importante en la patogénesis de la EA. En este artículo, revisamos el papel de KLF4 en la neuroprotección y la neurogénesis en la EA.
KLF4 no es solo un regulador de la regulación de la proliferación y diferenciación celular, sino también un objetivo potencial para regular las respuestas inmunitarias. KLF4 puede regular los factores inflamatorios negativos y promover la respuesta inflamatoria, y tener un gran efecto en la expresión de la microglía nuclear de astrocitos. Además, KLF4 y ERK5 pueden actuar juntos para ejercer acciones neuroprotectoras. Además, la regeneración de axones se puede lograr alterando el contenido de factores de transcripción específicos, inhibidores intracelulares o alterando las vías de señalización intracelular. Eliminar KLF4 puede mejorar la regeneración del axón y acelerar la tasa de crecimiento del axón. La reducción de la expresión de KLF4 promueve la regeneración de axones a través de la vía JAK-STAT3, y KLF4 promueve la vía JAK-STAT3 para una mayor regeneración de axones. Por lo tanto, KLF4 podría estar involucrado en el proceso de antiinflamatorio, antiapoptosis, regeneración de axones y acumulación de hierro en el SNC, que desempeña un papel fundamental en la generación de EA. Estos hallazgos sugieren que KLF4 representa una diana terapéutica potencial para la EA. Sin embargo, los mecanismos celulares y moleculares profundos de los efectos de KLF4 en la EA siguen sin estar claros y se necesitan más investigaciones.
Contribuciones de los autores
ZQC, XHZ e YJ escribieron el manuscrito. SHG y JYL modificaron el marco del manuscrito. BJL y RJC proporcionaron las revisiones críticas. Todos los autores aprobaron la versión final del manuscrito para su envío.
Financiación
Este trabajo fue apoyado por subvenciones de la Fundación de Ciencias Naturales de China (NSFC) (81871070, 31571126, 31471120) y financiación de la Agencia de Ciencia y Tecnología Jilin (Subvenciones Nos.20180519003JH, 20180414051GH, 20170414034GH y 20180414050GH).
Declaración de Conflicto de Intereses
Los autores declaran que la investigación se realizó en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un conflicto de intereses potencial.
Cacabelos, R. (2018). ¿Ha habido mejoras en el descubrimiento de medicamentos para la enfermedad de Alzheimer en los últimos 5 años? Exp. Opin. Drug Discov. 13, 523–538. doi: 10.1080/17460441.2018.1457645
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
Liu, Y., Shi, J., and Chen, X. (2015). Identificación de nuevas dianas para la rinitis alérgica estacional durante y fuera de la temporada de polen mediante análisis de microarrays. Acta Otorrinolaringol. 135, 1330–1336. doi: 10.3109 / 00016489.2015.1067906
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Serini, S., y Calviello, G. (2016). Reducción del estrés oxidativo/nitrosativo en el cerebro y su implicación en el efecto neuroprotector de los AGPI n-3 en la enfermedad de Alzheimer. Curr. Alzheimer Res. 13, 123-134. doi: 10.2174/1567205012666150921101147
Resumen de PubMed / Texto completo de CrossRef / Google Scholar