- Introducción
- Presentación del caso
- Materiales y Métodos
- Cariotipo y Matriz SNP
- Secuenciación de Exoma completo
- Extracción, PCR y secuenciación de ARN
- Resultados
- Discusión
- Disponibilidad de datos
- Declaración de Ética
- Contribuciones de los autores
- Financiación
- Declaración de Conflicto de Intereses
- Agradecimientos
- Material suplementario
Introducción
El gen de la molécula de adhesión celular L1 (L1CAM) es una molécula de adhesión celular neuronal perteneciente a la superfamilia de inmunoglobulinas; posee funciones clave en el desarrollo del sistema nervioso (Itoh y Fushiki, 2015). Las mutaciones en L1CAM se han relacionado con síndromes neurológicos ligados al cromosoma X, que se resumen como enfermedades L1. Se clasifican de la siguiente manera: Hidrocefalia ligada al cromosoma X (XLH) debida a estenosis del acueducto de Sílvia (HSA), síndrome de MASA (discapacidad intelectual, afasia, marcha arrastrada, pulgares aducidos), paraparesia espástica tipo 1 (SP1) y agenesia del cuerpo calloso ligada al cromosoma X (ACC) (Weller y Gartner, 2001; Itoh y Fushiki, 2015).
En HGMD® Professional 2019.2 (https://portal.biobase-international.com/hgmd/pro/all.php) se han notificado alrededor de 282 mutaciones causantes de enfermedad (DMs) en el gen L1CAM. Las alteraciones en el gen L1CAM son variadas; los análisis de datos de mutaciones de 282 pacientes revelaron 51% de mutaciones sin sentido y sin sentido, 25% de supresiones, 5% de inserciones y 19% de cambios en el sitio de empalme, pero las mutaciones silenciosas en L1CAM con potencial patógeno fueron raras, y las mutaciones silenciosas a menudo se ignoraron, especialmente en la detección de secuenciación de exoma completo (WES).
En este estudio, utilizando WES, se examinó el ADN fetal de una mujer embarazada china que ha reportado cinco embarazos continuos con hidrocefalia fetal; solo encontramos una nueva mutación silenciosa c. 453G > T (p. Gly151 = ) en el gen L1CAM. Curiosamente, a través de análisis adicionales, indicamos que la mutación silenciosa creó una secuencia potencial de consenso en el sitio de empalme de 5′, que resultaría en una deleción en el marco de 72 pb del exón 5 y 24 aminoácidos de la proteína L1CAM.
Presentación del caso
Una mujer sana de 28 años de edad fue remitida a nuestra clínica después de cuatro interrupciones voluntarias del embarazo por hidrocefalia fetal en otros hospitales. Todos los fetos eran varones. Al llegar a nuestro hospital (Hospital de Mujeres, Facultad de Medicina, Universidad de Zhejiang, Zhejiang, China), ya estaba en su quinto embarazo a las 24 semanas de gestación, con una hidrocefalia fetal por exámenes de imagen. Para explorar la causa genética, se realizó una muestra de sangre fetal a las 26 semanas de edad gestacional. Se realizaron estudios citogenéticos convencionales para muestras fetales y parentales, y la muestra fetal se analizó más a fondo mediante una matriz de polimorfismo de nucleótido único (SNP) y WES.
Este estudio se realizó de acuerdo con las recomendaciones del Comité de Ética del Hospital de Mujeres de la Facultad de Medicina de la Universidad de Zhejiang, y se obtuvo el consentimiento informado de todos los participantes de este estudio de acuerdo con la Declaración de Helsinki. El protocolo del estudio fue aprobado por la Junta de Revisión del Hospital de Mujeres, Facultad de Medicina, Universidad de Zhejiang en China.
Materiales y Métodos
Cariotipo y Matriz SNP
Los cariotipos de sangre del cordón fetal y sangre del cordón periférico se determinaron mediante cariotipo convencional de al menos 30 linfocitos sanguíneos, que fueron detenidos en metafase por colchicina. Los cariotipos de bandas G de células cultivadas se realizaron en el nivel de banda 320-400 con una resolución de alrededor de 10 Mb. La matriz SNP fue realizada por la matriz CytoScan ™ HD (Affymetrix, EE. UU.) de acuerdo con las instrucciones del fabricante, con alrededor de 2,600,000 marcadores, incluidos 750,000 sondas SNP y 1,900,000 sondas sin polimorfismo para una cobertura completa del genoma completo. Los datos se analizaron mediante el software CHAS (Chromosome Analysis Suite) (Affymetrix, Santa Clara, CA) basado en el ensamblaje GRCh37/hg19. El umbral de notificación del resultado del número de copias se fijó en 500 kb con un recuento de marcadores ≥50 para las ganancias y en 200 kb con un recuento de marcadores ≥50 para las pérdidas.
Secuenciación de Exoma completo
La parte principal de WES fue proporcionada por el Instituto de Genómica de Beijing. El ADN genómico fue extraído por un Kit de sangre DNeasy (Qiagen, CA) y luego fue fragmentado por Covaris LE220 (Massachusetts, EE.UU.) para generar una biblioteca de extremos emparejados (200-250 pb). Todas las bibliotecas amplificadas se realizaron en la plataforma BGISEQ – 500, el ADN de una sola hebra se mezcló con el Kit de Preparación de bibliotecas de ADN MGIEasy™ V1 (BGI, Shenzhen, China) y luego se secuenció utilizando química 100SR con el Kit de secuenciación de alto rendimiento BGISEQ-500RS (BGI, Shenzhen, China).
Las lecturas limpias (con una longitud de 90 pb) derivadas de la secuenciación y filtrado específicos se alinearon con la referencia del genoma humano (hg19) utilizando el paquete de software de visión múltiple Burrows-Wheeler Aligner (BWA) (Li y Durbin, 2009). Después de la alineación, los archivos de salida se utilizaron para realizar análisis de profundidad y cobertura de secuenciación de la región objetivo, variantes de nucleótido único (SNV) y llamadas indel, utilizamos el software GATK para detectar SNV e indel (McKenna et al., 2010), todos los SNV y los indel se filtraron y estimaron a través de múltiples bases de datos, incluida la Base de Datos de Polimorfismo de Nucleótido Único (dbSNP) del Centro Nacional de Información Biotecnológica (NCBI), el HapMap, el conjunto de datos del Proyecto 1000 Genomas y la base de datos de 100 adultos sanos chinos. Utilizamos Condel, TAMIZ, Polifen-2, RTL, Probador de mutaciones y Filop para predecir el efecto de las variantes. Las variantes patógenas se evalúan según el protocolo emitido por el American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015). Se utilizó la Base de Datos de Mutaciones Genéticas Humanas (HGMD, por sus siglas en inglés) para detectar mutaciones. Todas las variantes patógenas potenciales se validaron utilizando métodos de secuenciación de Sanger.
Extracción, PCR y secuenciación de ARN
Se aislaron células mononucleares de sangre periférica (PMBC) y células mononucleares de sangre del cordón umbilical (CBMC) mediante separación de gradiente de densidad Ficoll. El ARN total se extrajo de PMBCS y CBMCs usando TRIzol (Takara, Japón). Los ARN totales extraídos se transcribieron a la inversa utilizando el kit de RT (Takara, Japón). La PCR se realizó utilizando GoldStar Best Master Mix (CWBIO, Beijing). Se enumeran las secuencias de imprimación: L1CAM-DNA-5F, CCCACCCGTCCTTTTCCTA; L1CAM-DNA-5R, CGCTCGTCCTGTTGATGT; L1CAM-ARNm-4-6-F, GGTGTCCACTTCAAACCCAA; y L1CAM-ARNm-4-6-R, GCGGCTTCCTGTCAATCA. La secuenciación de Sanger se realizó mediante un analizador de ADN ABI 3130.
Resultados
Una mujer sana de 28 años de edad fue remitida a nuestra clínica después de cuatro interrupciones voluntarias del embarazo por hidrocefalia fetal. Todos los fetos eran varones (Figura 1A). El pedigrí familiar parecía mostrar XLH. Ya estaba en el quinto embarazo a las 26 semanas de gestación. La ventriculomegalia fetal se detectó mediante ecografía fetal y resonancia magnética, que demostraron consistentemente la presencia de hidrocefalia. Mostraron que el ventrículo cerebral bilateral y el tercer ventrículo estaban obviamente dilatados, y que había hidrocefalia severa en el intracerebral y agenesia del cuerpo calloso (Figura 1B).FIGURA
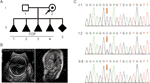
Figura 1 A) Genealogía de la familia. ARRIBA, interrupción del embarazo. B) Exámenes por imágenes del feto. La ecografía fetal y la resonancia magnética fetal mostraron que había hidrocefalia grave en el feto. C) Análisis de secuencias de ADN genómico de miembros de la familia. Los genotipos de L1CAM fueron de tipo salvaje, c. 453G > T Het, y c. 453G > T Hom, en I:1 (marido), I:2 (mujer embarazada) y II:5 (feto). La mutación está indicada por las flechas rojas.
Con el fin de explorar la posible causa genética, realizamos análisis de cariotipo y matriz SNP para analizar la muestra de sangre fetal y no encontramos hallazgos positivos. El resultado de la neovascularización coroidea (NVC) se ha depositado en el Omnibus de Expresión Génica (GEO); el número de adhesión es GSE133063, como se adjunta a continuación (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
Luego, detectamos el feto por WES. La estrategia analítica para encontrar la posible identificación de variantes patógenas se muestra en la Figura S1. Se obtuvo una lista de variantes (Tabla S1) mediante el cribado de frecuencias de variantes, estado de mutación y modo de herencia. Teniendo en cuenta los genes asociados a la hidrocefalia (HP:0000238, http://compbio.charite.de/hpoweb/showterm?id=HP:0000238#id=HP_0000238) (Tabla S2), no hubo ninguna mutación notable adicional, excepto la mutación silenciosa de c. 453G > T en el exón 5 del gen L1CAM (NM_000425.3). c. 453G > T no se notificó en HGMD y ClinVar y no se encontró en dbSNP, gnomAD y otros conjuntos de datos. De acuerdo con las normas y directrices de la ACMG (Richards et al., 2015), aún no había alcanzado el criterio de «patógeno» o «probable patógeno», pero no había otras mutaciones potenciales; no tuvimos más remedio que hacer un análisis más detallado de la mutación silenciosa encontrada.
De acuerdo con el pensamiento tradicional, esta sustitución de bases ocurrió en la tercera base en el codón 151, que codifica una glicina, creando así una mutación neutra (p. Gly151 =). Esta variante se confirmó en el ADN extraído de la sangre del cordón umbilical fetal y la sangre periférica de la pareja mediante secuenciación de Sanger (Figura 1C). La mujer portaba la mutación heterocigota, y su marido era un genotipo de tipo salvaje.
Con Catador de mutaciones (http://www.mutationtaster.org/), c. 453G > T se calificó como «causante de enfermedad».»Mostró que las características de las proteínas podrían verse afectadas y el sitio de empalme podría cambiar; teníamos curiosidad por los posibles efectos de empalme de la función L1CAM en esta mutación silenciosa. La mutación silenciosa se probó utilizando los siguientes productos de software en línea: NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) y NNSplice (http://www.fruitfly.org/seq_tools/splice.html); también se predijo que el sitio de empalme de 5 pies de potencial se crearía en el L1CAM c.453G > Mutación T utilizando los productos de software (Figura S2). Los resultados mostraron que esta mutación silenciosa creó un sitio de empalme potencial de 5′ a 72 pb aguas arriba del sitio de empalme normal de exón 6/intrón 6 (Figura 2A). Si este es el caso, podemos encontrar el cambio de longitud del ARN mensajero L1CAM (ARNm) entre I:2 y II:5 (Figura 2A). La RT-PCR se realizó utilizando cebadores diseñados para amplificar exones 4-6 en ARNm L1CAM. De hecho, los resultados mostraron una banda corta de truncamiento en la PCR de ADNc fetal (II: 5), mientras que la banda amplificada a partir del ARNm L1CAM contenía la banda larga esperada en la PCR de ADNc del marido (I:1) y bandas largas / cortas en la PCR de ADNc de la mujer embarazada (I:2) (Figura 2B). La secuenciación directa del fragmento amplificado mostró que la deleción involucró los últimos 72 pb del exón 5 en el ADNc fetal masculino (la mujer era portadora) (Figura 2C). Tenemos la evidencia patógena crucial.FIGURA
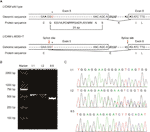
Figura 2 (A) Representación esquemática de la organización del exón 5, intrón 6 y exón 6 en L1CAM. B) Análisis por RT-PCR de los exones 5 y 6 del ADNc L1CAM de células mononucleares de sangre periférica (PMBC) y células mononucleares de sangre del cordón umbilical (CBMC). Electroforesis en gel de agarosa de productos RT-PCR generados a partir de I: 1 (marido), I:2 (mujer embarazada) y II:5 (feto). C) Análisis de secuencias del producto de RCP-RT a partir de CMBP de la pareja y CMBC del feto.
Esta mutación silenciosa resultó en 24 aminoácidos de la proteína L1CAM (residuos 151-174); Lys (K) fue sustituido por Glu (E) en el codón 175 (Figura 2A). Hubo alineación de múltiples secuencias de proteínas L1CAM en varias especies y conservación de los aminoácidos faltantes en L1CAM en mamíferos: Homo sapiens, Pan troglodytes, Bos taurus, Mus musculus y Rattus norvegicus (Figura 3A). Las proteínas L1CAM de mutación de empalme de tipo salvaje y c. 453G > T fueron predichas por el servidor de software CPHmodels-3.2 (http://www.cbs.dtu.dk/services/CPHmodels/) (Figura 3B). El dominio 2 similar a la inmunoglobulina (similar a la Ig) (residuos 134-230) de las proteínas L1CAM de mutación de tipo salvaje y de empalme se muestra en la Figura 3C. La mutación de empalme L1CAM c. 453G > T alteró la estructura de la proteína, especialmente el dominio 2 similar a la Ig

Figura 3 (A) Alineación de múltiples secuencias de proteínas L1CAM entre especies. El L1CAM c.453G > T dieron como resultado 24 aminoácidos de proteína L1CAM (residuos 151-174) faltantes en la región de aminoácidos conservados en diferentes especies. La columna negra muestra los aminoácidos que faltan. B) Las estructuras de la proteína L1CAM de mutación de empalme de tipo salvaje y c. 453G > T, según lo previsto por el servidor de software CPHmodels-3.2. C) Las estructuras del dominio 2 similar a la inmunoglobulina (Ig) (residuos 134-230) de la proteína L1CAM de mutación de tipo salvaje y de empalme.
Discusión
Las mutaciones silenciosas fueron a menudo detectadas por WES, pero no se ha prestado suficiente atención, lo que llevó a la omisión de DMs. En este estudio, empleamos a WES para explorar la causa genética de una familia china con hidrocefalia, pero solo encontramos una nueva mutación silenciosa en L1CAM, lo que nos obligó a hacer un análisis adicional. Afortunadamente, probamos que la mutación silenciosa creó un nuevo sitio de empalme de 5 pies y era un DM.
Las mutaciones en L1CAM pueden causar una enfermedad L1 ligada al cromosoma X, pero los síntomas clínicos son variables; las mutaciones producen fenotipos inesperados. En el estudio, los cinco fetos que sufren son todos varones, lo que es consistente con un patrón de herencia. La ecografía fetal y la resonancia magnética muestran una enfermedad L1 típica, que incluye XLH y agenesia del cuerpo calloso. Mejora nuestra comprensión de la correlación genotipo–fenotipo de L1CAM.
L1CAM c. 453G > T (p. Gly151 = ) se pensó inicialmente que no tenía ningún efecto sobre la secuencia de proteínas. Pero otras mutaciones silenciosas, c. 924C > T (p. Gly308 = ) y c. 645C > T (p. Gly215=), en el gen L1CAM han sido reportadas como DMs (Du et al., 1998; Vos et al., 2010). La c.La mutación 924C > T dio lugar a la activación de un nuevo sitio de empalme 69 pb 5′ al sitio de empalme normal del donante exón 8/intrón 8, y se ha declarado como un sitio «causante de enfermedad» para hidrocefalia (Du et al., 1998). Para c. 645C > T en L1CAM, se eliminó 51 pb con la activación de un nuevo empalme de donante exón 6 / intrón 6(Vos et al., 2010). Nuestro presente estudio fue similar; la mutación de c. 453G > T creó un sitio potencial de empalme de 5′ aguas arriba del sitio normal de empalme de exón 5/intrón 5. Todas estas mutaciones silenciosas crearon nuevos sitios de empalme de donantes, lo que resultó en que se omitiera el exón. Nos recordó que debemos prestar mucha atención a estas mutaciones silenciosas, que pueden afectar el empalme de proteínas.
Como glicoproteína transmembrana y miembro de la superfamilia de inmunoglobulinas de moléculas de adhesión celular, la proteína L1CAM puede interactuar en la superficie celular con una serie de glicoproteínas diferentes, y la unión homofílica es probablemente su principal modo de interacción (Wei y Ryu, 2012). Los estudios sobre la estructura cristalina de los dominios similares a Ig 1-4 en neurofascina sugirieron que muchas mutaciones patológicas de L1 afectan los residuos de aminoácidos conservados dentro de estos dominios e interfieren con las interacciones homofílicas (Liu et al., 2011), especialmente según lo verificado por la investigación de funciones del dominio 2 similar al Ig (Zhao et al., 1998). En nuestro estudio, especulamos que L1CAM c. 453G > T alteró el dominio 2 similar a Ig en la parte extracelular de la proteína L1CAM, lo que llevó a la interacción extracelular anormal, fallando en iniciar la vía de señalización aguas abajo. Un estudio adicional dedicado a la espectrometría de masas de esta variante L1CAM aclararía específicamente qué conjunto molecular se produce en la célula.
En resumen, a través de WES, reportamos una nueva mutación silenciosa c. 453G > T en L1CAM que produce un sitio de empalme de 5′ responsable de hidrocefalia. Se predijo que esta variante de proteína anormal alteraría el dominio 2 similar a Ig, lo que podría afectar la unión homofílica de la proteína L1CAM. Además, se realizó un diagnóstico genético prenatal para la mujer embarazada que reportó cinco embarazos continuos con hidrocefalia. Mientras tanto, sugirió que algunas mutaciones silenciosas detectadas en WES no deberían ignorarse; las predicciones de empalme de estas mutaciones eran necesarias. Proporcionó una nueva base genética para el diagnóstico prenatal y el diagnóstico prenatal previo a la implantación de la hidrocefalia.
Disponibilidad de datos
En este estudio se analizaron conjuntos de datos disponibles públicamente. Estos datos se pueden encontrar aquí: GSE133063 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
Declaración de Ética
Los estudios con participantes humanos fueron revisados y aprobados por el Comité de Revisión del Hospital de la Mujer, Facultad de Medicina, Universidad de Zhejiang en China. El consentimiento informado por escrito para participar en este estudio fue proporcionado por el tutor legal/familiar de los participantes. Se obtuvo el consentimiento informado por escrito de la(s) persona(s) y del (de los) tutor (es) legal (es) del menor (es)/familiar más cercano, para la publicación de cualquier imagen o dato potencialmente identificable incluido en este artículo.
Contribuciones de los autores
Experimentos conducidos por YS, YLi, MC, YLu, YQ e YY. YS preparó las cifras. MC e YLi analizaron los datos de WES. YLu y YQ realizaron análisis de cariotipo y matriz SNP. YY reclutó muestras. HL y FL proporcionaron exámenes por imágenes. YS y MD escribieron el manuscrito. Todos los autores leyeron y aprobaron el manuscrito final.
Financiación
Este estudio fue apoyado por la Fundación Nacional de Ciencias Naturales de China (Subvención No.81801441), el Programa Clave de Investigación y Desarrollo de la provincia de Zhejiang (Subvención No. 2019C03025), el Programa de Investigación y Desarrollo Clave Nacional de China (Subvención No.2016YFC1000703) y la Fundación de Investigación Científica Médica de la provincia de Zhejiang (Subvención No. 2014KYA246).
Declaración de Conflicto de Intereses
Los autores declaran que la investigación se realizó en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un conflicto de intereses potencial.
Agradecimientos
Agradecemos a los pacientes inscritos en esta investigación. Agradecemos al Dr. Jiong Gao (BGI Genomics, BGI-Shenzhen, Shenzhen 518083, China) por su asistencia durante la preparación de este manuscrito.
Material suplementario
El Material Suplementario para este artículo se puede encontrar en línea en: https://www.frontiersin.org/articles/10.3389/fgene.2019.00817/full#supplementary-material
Figura S1 / Estrategia analítica para encontrar la posible identificación de variantes patógenas por WES.
Figura S2 / Sitios de empalme de donantes predichos por NetGene2 y NNSplice.
Tabla S1 / Lista de variantes mediante el cribado de frecuencias de variantes, estado de mutación y modo de herencia.
Cuadro S2 / Lista de genes asociados a hidrocefalia (Exportación para HP:0000238).
Du, Y. Z., Dickerson, C., Aylsworth, A. S., Schwartz, C. E. (1998). Una mutación silenciosa, C924T (G308G), en el gen L1CAM produce hidrocefalia ligada al cromosoma X (HSA). J. Med. Genet. 35 (6), 456–462. doi: 10.1136 / jmg.35.6.456
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Itoh, K., Fushiki, S. (2015). El papel de la L1cam en la corticogénesis murina y la patogénesis de la hidrocefalia. Pathol. Int. 65 (2), 58–66. doi: 10.1111 / pin.12245
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Li, H., Durbin, R. (2009). Alineación rápida y precisa de lectura corta con la transformación Burrows-Wheeler. Bioinformática 25 (14), 1754-1760. doi: 10.1093 / bioinformática / btp324
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Liu, H., Focia, P. J., He, X. (2011). Mecanismo de adhesión homofílica de la neurofascina, un miembro de la familia L1 de moléculas de adhesión de células neuronales. J. Biol. Chem. 286 (1), 797–805. doi: 10.1074 / jbc.M110.180281
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data (en inglés). Genome Res. 20 (9), 1297-1303. doi: 10.1101 / gr.107524.110
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Mediterráneo. 17 (5), 405–424. doi: 10.1038 / gim.2015.30
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Vos, Y. J., de Walle, H. E. K., Bos, K. K., Stegeman, J. A., Berge, A. M., Bruining, M., et al. (2010). Correlaciones genotipo-fenotipo en el síndrome L1: una guía para el asesoramiento genético y el análisis de mutaciones. J. Med. Genet. 47 (3), 169–175. doi: 10.1136 / jmg.2009.071688
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Wei, C. H., Ryu, S. E. (2012). Interacción homofílica de la familia L1 de moléculas de adhesión celular. Exp. Mol. Mediterráneo. 44 (7), 413–423. doi: 10.3858 / emm.2012.44.7.050
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Weller, S., Gartner, J. (2001). Aspectos genéticos y clínicos de la hidrocefalia ligada al cromosoma X (enfermedad L1): mutaciones en el gen L1CAM. Zumbido. Mutat. 18 (1), 1–12. doi: 10.1002 / humu.1144
PubMed Abstract | CrossRef Full Text | Google Scholar
Zhao, X., Yip, P. M., Siu, C. H. (1998). Identification of a homophilic binding site in immunoglobulin-like domain 2 of the cell adhesion molecule L1. J. Neurochem. 71 (3), 960–971 doi: 10.1046/j.1471-4159.1998.71030960.x.
PubMed Abstract | CrossRef Full Text | Google Scholar