La tecnología para la secuenciación de ADN se desarrolló en 1977 gracias a Frederick Sanger. Tomó un poco más de tiempo antes de que fuera posible secuenciar un genoma completo. Esto se debe a que necesitábamos un modelo matemático apropiado y un poder computacional masivo para ensamblar millones o miles de millones de lecturas pequeñas en un genoma completo más grande. La potencia computacional y el software de hoy en día son la principal diferencia entre lo que solía llevar años de trabajo a principios de la década de 2000 y lo que solo toma unas pocas horas hoy en día. El algoritmo que eligió para hacer esto es el «santo grial» de la tecnología de ensamblaje. Estos algoritmos incorporan una de las variables más famosas conocidas en los modelos matemáticos, el k-mer.
El origen del k-mer y el modelo matemático que lo rodea proviene de un matemático suizo de 1735, Leonhard Euler, conocido como el padre de la función matemática. Un matemático holandés Nicolaas de Bruijn adaptó las ideas de Euler para encontrar una secuencia cíclica de letras tomadas de un alfabeto dado para el que cada palabra posible de cierta longitud aparece como una cadena de caracteres consecutivos en la secuencia cíclica exactamente una vez.
el algoritmo de de Bruijn fue adaptado por biólogos moleculares, que muchos años después se enfrentaron a un problema equivalente: cómo ensamblar secuencias de ADN. Por lo tanto, los científicos de todo el mundo ahora utilizan el gráfico De Bruijn y la variable k.
Aplicación de k-mers para Ensamblar Secuencias de ADN
En pocas palabras, el ensamblaje del genoma de novo implica conectar lecturas de ADN pequeñas consecutivas y terminar con secuencias más grandes. Para generar un gráfico de Bruijn (ver la figura a continuación), los nucleótidos en el borde de cada lectura deben superponerse al borde de un segundo (y así sucesivamente). El objetivo final es crear un vértice consecutivo, que (potencialmente) resultará en grandes fragmentos de ADN.
Tienes que fragmentar tus lecturas en k-mers, que son un número específico de nucleótidos que se superponen. El k-mer le permite generar una secuencia única a partir de muchos pequeños. Se identifica cada secuencia única de k-mer y se eliminan copias adicionales. Este aspecto de k-mers le permite superar uno de los inconvenientes de la secuenciación de próxima generación: obtener lecturas que representan regiones genómicas con diferentes frecuencias (es decir, obtener muchas lecturas pequeñas de una región). El uso de k-mers elimina las secuencias repetidas más de una vez debido a la cobertura desigual de las secuencias. Sin embargo, tenga en cuenta que un tamaño bajo de k-mer aumentará las posibilidades de que los nucleótidos se superpongan, mientras que tener un valor más grande los disminuirá.
La tecnología de ensamblaje de novo de hoy en día es más eficiente cuando se utilizan bibliotecas de lecturas grandes (es decir, 1.000–10.000 bps) combinadas con bibliotecas más pequeñas (100-200 bps). Los programas de software pueden usar el valor k y k-mers para ensamblar lecturas cortas. Estos pueden ser incorporados y verificados por otros más grandes para terminar en contigs más precisos.
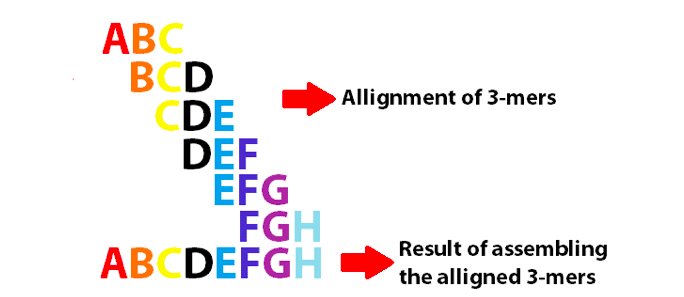
de un gráfico de Bruijn que utiliza 3 mers para ensamblar las 8 primeras letras del alfabeto inglés. Tenga en cuenta que estos 3 mers se superponen como k-1.
Cuanto Más Sepa, Más Podrá Lograr en el Ensamblaje de ADN
Hay consejos específicos que debe considerar antes de aplicar los gráficos De De Bruijn en su método de ensamblaje y elegir el tamaño k-mer más apropiado. Al aprovechar estos, puede generar mejores resultados.
- En primer lugar, y tal vez lo más importante, es usar muchos k-mers diferentes en su ensamblaje. A continuación, debe evaluar sus resultados y elegir el mejor(s). Nunca olvides que casi nunca hay un solo ensamblaje correcto.
- Debe manejar cuidadosamente las lecturas de errores antes de usar un k-mer. Si no elimina cuidadosamente los errores, los resultados pueden crear un bulto no deseado, complicando su ensamblaje. Aumente el umbral de la tasa de error que utiliza durante el recorte de secuencias. Puede que pierdas algunas secuencias, pero las que queden serán las mejores.
- Debe manejar cuidadosamente las repeticiones de ADN. Por ejemplo, la secuenciación de Illumina genera una gran cantidad de datos. Primero, intente ensamblar una pequeña fracción de las lecturas y luego utilícelas todas para detectar diferencias. Las lecturas cortas repetibles pueden interferir negativamente con el proceso de ensamblaje.
- Conozca sus datos. Si no conoce el tamaño de su genoma esperado, la cantidad de cobertura de secuenciación y el número de lecturas, entonces es más propenso a elegir el mejor valor k para ensamblar su genoma. Puedes visitar k-mer advisors, como velvet advisor de la universidad de Monash para obtener algunos consejos sobre qué valor parece más adecuado.
El uso de k-mers de varias longitudes y la alineación de los contig también ayuda a los investigadores a detectar las tasas de mutación, ampliando su uso. Por supuesto, manipular los gráficos de De Bruijn hacia el beneficio del ensamblaje no es una panacea. Hay muchas cosas a considerar además de una función simplista para ensamblar el genoma de un organismo vivo. Esto es solo una introducción de la historia y de cómo los biólogos pueden usarla de manera más eficiente.
- Compeau PE, Pevzner PA, Tesler G. (2011). Cómo aplicar los gráficos de Bruijn al ensamblaje del genoma.Biotecnología de la Naturaleza. 29(11):987–91.
- Aggarwala V, Voight BF. (2016). Un modelo de contexto de secuencia expandida explica ampliamente la variabilidad en los niveles de polimorfismo en todo el genoma humano. Genética de la Naturaleza. 48(4): 349–55.

¿Esto le ha ayudado? Entonces, por favor, comparta con su red.