DNA-sekvensointitekniikka kehitettiin jo vuonna 1977 Frederick Sangerin ansiosta. Kesti hieman kauemmin ennen kuin oli mahdollista sekvensoida täydellinen genomi. Tämä johtuu siitä, että tarvitsimme sopivan matemaattisen mallin ja massiivisen laskentatehon kootaksemme miljoonia tai miljardeja pieniä lukuja suurempaan kokonaiseen genomiin. Nykypäivän laskentateho ja ohjelmistot ovat suurin ero siihen, mikä ennen vei vuosia työtä 2000-luvun alussa ja mikä kestää nykyään vain muutaman tunnin. Valitsemasi algoritmi on kokoamisteknologian Graalin malja. Nämä algoritmit sisältävät yhden kuuluisimmista matemaattisissa malleissa tunnetuista muuttujista, k-mer: n.
k-Merin ja sitä ympäröivän matemaattisen mallin alkuperä on peräisin vuonna 1735 syntyneeltä sveitsiläiseltä matemaatikolta Leonhard Eulerilta, joka tunnetaan matemaattisen funktion isänä. Hollantilainen matemaatikko Nicolaas de Bruijn muokkasi Eulerin ajatuksia löytääkseen syklisen kirjainjonon, joka on otettu tietystä aakkostosta ja jossa jokainen mahdollinen tietyn pituinen sana esiintyy peräkkäisten merkkien jonona syklisessä järjestyksessä tasan kerran.
de Bruijnin algoritmia muokkasivat molekyylibiologit, jotka vuosia myöhemmin kohtasivat vastaavan ongelman: miten DNA-sekvenssit kootaan. Niinpä tiedemiehet kaikkialla maailmassa käyttävät nyt De Bruijnin graafia ja muuttujaa k.
K-mers: n soveltaminen DNA-sekvenssien kokoamiseen
muutamalla sanalla de Novon genomikokoonpanossa yhdistetään peräkkäiset pienet DNA-lukemat ja päädytään suurempiin sekvensseihin. Luodakseen de Bruijn-graafin (katso kuva alla), jokaisen lukeman reunalla olevien nukleotidien on oltava päällekkäin toisen (ja niin edelleen) reunan kanssa. Lopullinen tavoite on luoda peräkkäinen huippupiste, joka (mahdollisesti) johtaa suuriin DNA-fragmentteihin.
lukema pitää pirstoa k-mereiksi, jotka ovat tietty määrä nukleotideja, jotka menevät päällekkäin. K-mer avulla voit luoda ainutlaatuisen sekvenssin monista pienistä. Jokainen ainutlaatuinen k-mer sekvenssi tunnistetaan ja ylimääräiset kopiot poistetaan. Tämä näkökohta k-mers voit voittaa yksi haittoja seuraavan sukupolven sekvensointi-saada lukee, jotka edustavat genomialueita eri taajuuksilla (eli saada paljon pieniä lukee yhdeltä alueelta). K-Mersin käyttö eliminoi useammin kuin kerran toistuvat sekvenssit epätasa-arvoisen sekvenssikattavuuden vuoksi. Muista kuitenkin, että pieni k-mer-koko lisää nukleotidien päällekkäisyysmahdollisuuksia, kun taas suurempi arvo vähentää niitä.
nykypäivän de novo assembly-tekniikka on tehokkaampaa, kun käytetään kirjastoja, joissa on suuret lukemat (eli 1 000–10 000 peruspistettä) yhdistettynä pienempiin (100-200 peruspistettä). Ohjelmistot voivat käyttää K-arvoa ja k-mersiä lyhyiden lukujen kokoamiseen. Nämä voidaan sitten sisällyttää ja todentaa suuremmilla päätyä tarkempiin kontigs.
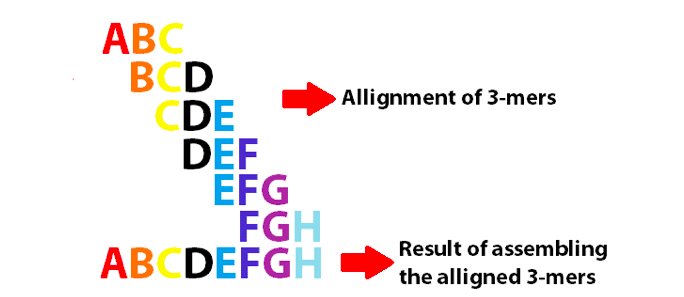
esimerkki de Bruijnin graafista, jossa koottiin 3-mers-kirjaimella Englannin aakkosten 8 ensimmäistä kirjainta. Huomaa, että nämä 3-Merit limittyvät K-1: ksi.
mitä enemmän tiedät, sitä enemmän voit saavuttaa DNA: n kokoonpanossa
on olemassa erityisiä vinkkejä, jotka sinun on otettava huomioon ennen kuin sovellat De Bruijn-kaavioita kokoonpanomenetelmässäsi ja valitset sopivimman k-mer-koon. Näitä hyödyntämällä voi saada aikaan parempia tuloksia.
- Ensinnäkin ja ehkä kaikkein tärkeintä on käyttää kokoonpanossaan monia erilaisia k-merejä. Sinun pitäisi sitten arvioida tuloksia ja valita paras(s). Älä koskaan unohda, että on lähes koskaan yksi ja vain yksi oikea kokoonpano.
- sinun tulee käsitellä huolellisesti virhelukemat, ennen kuin käytät k-mer-merkkiä. Jos et huolellisesti poistaa virheitä, tulokset voivat luoda ei-toivottuja pullistuma, vaikeuttaa kokoonpano. Korota kynnystä virhetasolle, jota käytät sekvenssin trimmauksen aikana. Saatat menettää joitakin sekvenssejä, mutta ne, jotka jäävät, ovat hienoimpia.
- sinun tulee käsitellä huolellisesti DNA-toistoja. Esimerkiksi Illumina-sekvensointi tuottaa hyvin suuren määrän dataa. Yritä ensin koota pieni murto-osa lukemista ja sitten käyttää niitä kaikkia havaitaksesi eroja. Toistettavat lyhyet lukemat saattavat häiritä negatiivisesti kokoamisprosessiasi.
- tunne tietosi. Jos et tiedä koko odotetun genomin, määrä sekvensointi kattavuus, ja määrä lukee, niin olet alttiimpi valita paras K arvo kokoamista genomin. Voit käydä k-mer advisors, kuten velvet advisor Monash university saada joitakin neuvoja siitä, mikä arvo tuntuu sopivampi.
eripituisten k-merien käyttäminen ja kontigien yhdenmukaistaminen auttaa myös tutkijoita havaitsemaan mutaationopeudet ja laajentaa sen käyttöä. De Bruijnin kaavioiden manipulointi kokoonpanohyötyä kohti ei tietenkään ole ihmelääke. On monia asioita, jotka on otettava huomioon kuin pelkistetty funktio elävän organismin genomin kokoamiseksi. Tämä on vain esittely historiasta ja siitä, miten biologit voivat käyttää sitä tehokkaammin.
- Compeau PE, Pevzner PA, Tesler G. (2011). Miten soveltaa de Bruijn kaavioita genomin kokoonpano.Luontobioteknologia. 29(11):987–91.
- Aggarwala V, Voight BF. (2016). Laajennettu sekvenssikontekstimalli selittää laajasti polymorfismitasojen vaihtelua ihmisen perimässä. Luonnongenetiikka. 48(4): 349–55.

onko tämä auttanut sinua? Jaa sitten verkkosi kanssa.