- Introduction
- Les caractéristiques biologiques de KLF4
- Rôle de KLF4 dans la MA
- Rôle de KLF4 dans la neuroinflammation
- Rôle de KLF4 dans l’apoptose
- Rôle de KLF4 dans la régénération des axones
- Rôle de KLF4 dans l’accumulation de fer
- Conclusion
- Contributions de l’auteur
- Financement
- Déclaration sur les conflits d’intérêts
Introduction
Le facteur 4 de type Kruppel (KLF4) est un membre de la famille des facteurs de transcription à doigt de zinc, qui est exprimé dans divers tissus humains. Il est bien connu comme l’un des quatre facteurs d’induction aux cellules souches pluripotentes (IPSC) (Ghaleb et Yang, 2017). KLF4 peut réguler plusieurs processus biologiques importants tels que la neuroinflammation, le stress oxydatif, la prolifération, la différenciation et l’apoptose (Kaushik et al., 2010; Mamonkin et coll., 2013; Zhang et coll., 2015; Miao et coll., 2017; Xu et coll., 2017). Des études antérieures portaient sur le rôle de KLF4 dans le développement et la progression du cancer (Karam et al., 2017; Yadav et coll., 2018). KLF4 est un facteur de transcription à double fonction, qui peut exercer son rôle d’oncogène ou de gène suppresseur de tumeur selon le type de cancer ou le stade du cancer (Evans et Liu, 2008). Il peut activer ou inhiber la transcription de gènes impliqués dans la prolifération, la différenciation et l’apoptose des cellules (Ding et al., 2015). KLF4 peut collaborer avec d’autres facteurs de reprogrammation pour convertir les cellules somatiques en IPSC et inhiber la différenciation des cellules souches (Takahashi et Yamanaka, 2006; van Schaijik et al., 2018). Cela offre des perspectives thérapeutiques pour les maladies vasculaires, les maladies immunitaires, l’anorexie et d’autres maladies (Imbernon et al., 2014; Liu Y. et coll., 2015; Murgai et coll., 2017). De plus, KLF4 peut jouer un rôle largement régulateur dans le système nerveux central (SNC). Plusieurs études indiquent que KLF4 est lié à de multiples troubles neurologiques, notamment la maladie d’Alzheimer (MA), l’épilepsie, la maladie de Parkinson, l’hydrocéphalie et la schizophrénie (Qin et al., 2011; Xie et coll., 2013; Han et coll., 2015; Nishiguchi et coll., 2015; Li L. et coll., 2017).
La MA est l’une des maladies neurodégénératives chroniques les plus courantes, qui entraîne des troubles cognitifs et de la mémoire, divers symptômes mentaux et des anomalies comportementales et la démence progressive est la caractéristique clinique la plus courante (Jiang et al., 2018). Les facteurs pathogènes confirmés actuels de la MA comprennent la formation de plaques séniles induites par un dépôt anormal d’amyloïde-β (Aß) et les enchevêtrements neurofibrillaires ou névrite dystrophique induits par l’accumulation de tau (Querfurth et LaFerla, 2010; Shinohara et al., 2014). De plus, la MA peut également être affectée par des facteurs génétiques. Cependant, la pathogenèse provoquée est encore obscure. Les médicaments les plus répandus pour le traitement de la MA comprennent les exhausteurs de neurotransmetteurs, les agents anti-amyloïdes, les peptides neuroprotecteurs et d’autres médicaments (Cacabelos, 2018). Notamment, plusieurs études ont montré que KLF4 a joué un rôle important dans la pathogenèse de la MA. Dans cette revue, nous nous concentrons sur le rôle régulateur de KLF4 dans la neuroinflammation, l’apoptose neuronale, la régénération axonale et l’accumulation de fer pour expliquer l’association entre KLF4 et la pathogenèse de la MA, ce qui pourrait fournir des informations sur les mécanismes cellulaires et moléculaires des troubles neurodégénératifs.
Les caractéristiques biologiques de KLF4
KLF4 est une protéine nucléaire contenant un doigt de zinc, isolée de la banque NIH 3T3 et située dans le noyau cellulaire. Il a d’abord été identifié et caractérisé par Shields et al. (1996). La masse moléculaire du KLF4 humain est de 55kD et il est situé sur le chromosome 9q31. KLF4 couvre un segment de gène de 6,3 kb et possède cinq exons. Sa région codante pour l’ADNc code pour un polypeptide constitué de 470 résidus d’acides aminés (Yet et al., 1998; Ghaleb et Yang, 2017). L’extrémité carboxy de KLF4 a une région de structure de liaison à l’ADN contenant trois structures de doigts de zinc de type Cys2His2 (C2H2), qui sont formées par 81 acides aminés hautement conservés. Il régule la transcription par affinité élevée avec les éléments CACCC et les séquences d’ADN de gènes cibles riches en GC (Shields et Yang, 1998; Pearson et al., 2008). La plupart des sites de liaison à l’ADN de KLF4 sont situés dans la région du doigt de zinc, y compris le domaine d’activation de la transcription N-terminale pour les protéines interagissant, la structure du doigt de zinc C-terminal pour la liaison à l’ADN et la zone d’inhibition de la transcription (Bieker, 2001). KLF4 est impliqué dans la régulation de l’expression de nombreux gènes endogènes (Shields et Yang, 1998). Il existe un domaine de régulation transcriptionnel très variable à l’extrémité amino de KLF4. Les résidus d’acides aminés situés entre le 91 et le 117 amino constituent un domaine d’activation transcriptionnelle, riche en proline et en sérine, tandis qu’un domaine de répression transcriptionnelle existe également. Par conséquent, KLF4 a deux effets indésirables: l’activation et l’inhibition de la transcription des gènes (Yet et al., 1998; Wei et coll., 2006).
Au cours du développement embryonnaire, KLF4 était plus élevé exprimé à la fin du développement embryonnaire. Alors que dans les tissus et organes matures, KLF4 est principalement exprimé dans le tractus gastro-intestinal, la cavité buccale, l’épiderme cutané, l’endothélium vasculaire et les reins, et est moins exprimé dans le cerveau (Segre et al., 1999; Ghaleb et coll., 2011; Liu et coll., 2013; Chen et coll., 2015; He et coll., 2015; Bin et coll., 2016). On pense qu’il joue un rôle important dans la régulation de la prolifération et de la différenciation cellulaires. En outre, KLF4 peut également réguler le cycle cellulaire. KLF4 peut activer P21 d’une manière dépendante de P53 (Zhang et al., 2000). De plus, il a été constaté que les cellules KLF4 (–/–) entraient en phase de sénescence plus tôt que les cellules KLF4 (+/+), ce qui peut s’expliquer par une expression moins antioxydante des gènes et un niveau plus élevé d’espèces réactives de l’oxygène (ROS) dans les cellules KLF4 (–/–). Le ROS peut augmenter l’expression de p53 et de p21 et favoriser par la suite les dommages à l’ADN (Liu C. et al., 2015). Il a été constaté que PRMT5 peut élever l’expression de KLF4 dans les niveaux de protéines. Il a été rapporté que PRMT5 augmentait la transcription de p21 et diminuait l’expression de bax via l’inhibition de l’ubiquitylation de KLF4 (Hu et al., 2015). De plus, de nombreuses études ont démontré que KLF4 est impliqué dans la régulation de l’apoptose des neurones (Kong et al., 2016; Cui et coll., 2017; Song et coll., 2018). Le rôle de régulation physiologique de KLF4 que nous connaissons est encore peu important et des investigations supplémentaires sont nécessaires.
Rôle de KLF4 dans la MA
Il est bien établi que la MA est principalement caractérisée par des troubles de la mémoire et des troubles cognitifs et un dysfonctionnement exécutif (Goedert et Spillantini, 2006). De nombreuses études ont démontré que l’apoptose neuronale et le dysfonctionnement synaptique sont à la base pathologique du déclin de la fonction cognitive (Caccamo et al., 2017; Guo et coll., 2017; Yoon et coll., 2018). Les dommages accumulés par le dépôt d’Aß, le stress oxydatif et l’accumulation de fer peuvent entraîner un dysfonctionnement neuronal et une apoptose des patients atteints de MA. Plusieurs études ont montré que le rôle régulateur de KLF4 semble être crucial dans le SNC. Considérant que KLF4 régule l’apoptose neuronale, la régénération synaptique, le stress oxydatif et la neuroinflammation, la relation entre KLF4 et la pathogenèse de la MA pourrait être une nouvelle cible potentielle pour le traitement de la MA.
Rôle de KLF4 dans la neuroinflammation
Des quantités d’études cliniques ont montré que l’Aß peut s’agréger et est le composant principal des dépôts extracellulaires du tissu cérébral des patients atteints de MA, ce qui peut altérer les synapses et les neurones environnants et entraîner la mort neuronale. Une sécrétion anormale ou une production excessive d’Aß entraîne des changements pathologiques de la MA, de sorte que le dépôt d’Aß est le lien central de la MA (Rajmohan et Reddy, 2017). De plus, des études ont montré qu’un dépôt excessif d’Aß peut stimuler les cellules gliales à sécréter des ROS et d’autres facteurs d’influence, entraînant un stress oxydatif. On savait que le stress oxydatif pouvait stimuler la production d’Aß. Par conséquent, l’Aß et le stress oxydatif peuvent interagir l’un avec l’autre et affecter la progression de la MA (Cheignon et al., 2018).
KLF4 a été rapporté comme modulateur potentiel et a un grand effet sur l’inflammation en médiant les macrophages et les cellules endothéliales (figure 1) (Yoshida et al., 2014; Kapoor et coll., 2015; Yang et coll., 2018). Dans le SNC, des réactions inflammatoires excessives et chroniques peuvent endommager le neurone et le neurogliocyte. Il a récemment été démontré que l’expression de KLF4 était en corrélation positive avec la neuroinflammation induite par l’Aß42. Dans les cellules microgliales BV2, l’oligomère Aß42 peut augmenter l’expression de KLF4, qui est médiée par P53 activé (Li L. et al., 2017). Dans des conditions inflammatoires, telles que l’accumulation d’Aß, la libération de cytokines pro-inflammatoires peut être stimulée lors de la génération de MA (Griffin et Barger, 2010). Le pouvoir neurotoxique et le pouvoir pro-inflammatoire des oligomères Aß42 solubles sont relativement plus élevés que le dépôt de fibres insolubles (Selkoe, 1991; Weinberg et al., 2018). Le silence de KLF4 est capable de restaurer la neuroinflammation médiée par Aß42, et la surexpression de KLF4 peut exacerber la neuroinflammation médiée par Aß42 (Li L. et al., 2017). L’accumulation d’Aß induit l’activation des astrocytes et des microglies (Rodríguez et al., 2016). Les astrocytes activés peuvent améliorer la neuroinflammation en libérant des facteurs pro-inflammatoires tels que l’IL-1, l’IL-6 et le TNF-α (Rubio-Perez et Morillas-Ruiz, 2012; Doméné et al., 2016). Le cercle vicieux des réponses inflammatoires conduit finalement à un dysfonctionnement et à une apoptose neuronale.
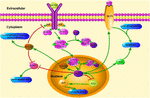
FIGURE 1. Illustration schématique des voies de signalisation liées à KLF4. Cette figure met en évidence le rôle de KLF4 dans la neuroprotection et la régénération des axones. Les flèches de la figure indiquent l’activation ou la promotion, et les lignes droites indiquent l’inhibition associée. KLF4, Facteur de type Kruppel 4; STAT3, Transducteur de signal et activateur de la transcription 3; JAK, Janus Kinase; SOCS3, Suppresseur de la signalisation des cytokines 3; HCP1, protéine porteuse de l’hème 1; ERK5, protéine kinase activée par les mitogènes 5.
KLF4 joue un rôle crucial dans la régulation des signaux pro-inflammatoires. Dans les cellules gliales, l’activation KLF4 induite par le gemfibrozil augmente le suppresseur du signal cytokine 3 (SOCS3) via la voie PI3-kinase-AKT (Ghosh et Pahan, 2012). Le knockdown médié par le siRNA de KLF4 pourrait atténuer le niveau de SOC dans l’astroglie et la microglie de souris, ce qui pourrait affecter par la suite l’expression du gène inflammatoire (Kaushik et al., 2010; Ghosh et Pahan, 2012). De plus, la délétion des SOC peut favoriser la survie des neurones blessés et favoriser la régénération des axones (Smith et al., 2009; Sun et coll., 2011). Et KLF4 régule positivement la production d’IL-1β ou d’autres marqueurs pro-inflammatoires. Il régule positivement la cyclooxygénase-2 (Cox-2) et régule négativement l’oxyde nitrique synthase inductible (iNOS) (Kaushik et al., 2013). De plus, KLF4 est un facteur de régulation important pour la différenciation des monocytes et une cible potentielle pour la régulation immunitaire (Alder et al., 2008). Par conséquent, KLF4 pourrait favoriser la neuroinflammation en régulant ces régulateurs négatifs.
Il convient de mentionner que dans le modèle de la maladie de Parkinson, KLF4 peut favoriser le stress oxydatif et la neurotoxicité induits par le MPP +, puis augmenter l’apoptose neuronale et retarder la prolifération cellulaire (Chen et al., 2013). Le stress oxydatif est un déséquilibre entre la peroxydation et l’antioxydation. Les radicaux libres peuvent provoquer des changements dans différentes macromolécules, entraînant des dommages cellulaires, un vieillissement cellulaire et des dommages tissulaires (Parajuli et al., 2013; Nie et coll., 2015). Le stress oxydatif peut aggraver l’inflammation précoce et la production d’Aß, puis aggraver la MA (Cai et al., 2011). Par conséquent, KLF4 peut être impliqué dans le stress oxydatif dans la MA.
Ces résultats impliquent que KLF4 joue un rôle clé dans la médiation de la neuroinflammation en activant la microglie et, par conséquent, la libération de cytokines pro-inflammatoires. Il a le potentiel d’améliorer la neuroinflammation. Jusqu’à présent, de nombreuses études sur la pathogenèse de la MA se sont concentrées sur la neuroinflammation. En tant que cible potentielle de régulation immunitaire, KLF4 peut favoriser les réponses inflammatoires de la microglie en affectant les régulateurs négatifs associés, ce qui a un grand effet sur le développement de la MA.
Rôle de KLF4 dans l’apoptose
Les changements neurodégénératifs comprennent la perte progressive des neurones et des synapses dans les régions cérébrales représentatives, telles que le cortex cérébral, l’hippocampe et d’autres régions sous-corticales. Les déficiences fonctionnelles du SNC induites par la perte neuronale sont permanentes (Citron, 2010). Un stress oxydatif soutenu peut entraîner une apoptose neuronale (Wu et al., 2010). Un grand nombre d’études ont confirmé que la MA est étroitement liée au stress oxydatif (Lee et al., 2012; Yui et coll., 2015). Il a été constaté que le stress oxydatif chronique peut améliorer l’expression de la Phospholipase A2 groupe 3 (Pla2g3) dans les astrocytes et perturber l’équilibre de l’Aß, et par conséquent conduire au développement de l’AD (Yui et al., 2015).
De nombreuses études ont démontré que KLF4 joue un rôle important dans l’inhibition du développement du stress oxydatif (Shi et al., 2014; Liu C. et coll., 2015). Il a été constaté que KLF4 peut favoriser l’apoptose des cellules induite par H2O2, cette action étant probablement causée par une augmentation de l’expression de bax et une diminution de l’expression de bcl-2 (Li et al., 2010). La quercétine pourrait réduire l’expression de KLF4 dans les cellules humaines de neuroblastome SH-SY5Y et augmenter l’expression du rapport bcl-2 / bax. De plus, la quercétine peut modérer le taux d’apoptose des cellules SH-5YSY et réduire l’activité enzymatique de la caspase-3 (Xi et al., 2012). Une étude récente a étudié l’effet neuroprotecteur de la protéine kinase 5 activée par le mitogène (MAP) (ERK5) contre le stress oxydatif. L’activation d’ERK5 peut réduire partiellement la mort des neurones de l’hippocampe induite par H2O2 et augmenter la neuroprotection induite par le NGF et le PC (Su et al., 2014). Nils et coll. utilisé un mutant de MEK5 (MEK5D) pour étudier la transcription activée par ERK5 et les réponses fonctionnelles dans les cellules endothéliales humaines, et identifié KLF4 était une nouvelle cible ERK5 en aval (Ohnesorge et al., 2010). Il a été constaté que la surexpression de KLF4 peut supprimer les réponses inflammatoires médiées par le TNF et réduire l’adhésion des leucocytes et l’apoptose basocellulaire. Ces résultats confirment que KLF4 possède des propriétés anti-inflammatoires et anti-apoptotiques (Ohnesorge et al., 2010). Des expériences ultérieures ont démontré que la disparition de la malformation caverneuse cérébrale 1 (CCM1) dans les cellules endothéliales active ERK5 via la voie du signal MEKK3-MEK5 et augmente l’expression de KLF4 (Cuttano et al., 2016). ERK5 joue un rôle de médiateur dans le préconditionnement (PC) et le facteur de croissance nerveuse (NGF) a régulé l’expression de KLF4 (Su et al., 2014). En outre, la neutralisation de KLF4 par ARNi peut également réduire la neuroprotection induite par le NGF ou le PC. La surexpression de KLF4 conduit à un rapport bcl-2/bax plus élevé dans les cellules stressées par H2O2 (Su et al., 2014). KLF4 surexprimé accélère les changements dans bcl-2 et bax en se combinant avec son promoteur correspondant (Li et al., 2010). La cascade ERK5 / KLF4 peut agir comme un pivot dans diverses voies qui protègent les neurones de la mort induite par le stress oxydatif (Su et al., 2014).
Le stress oxydatif a été considéré comme étroitement lié à de nombreuses maladies dégénératives. KLF4 joue un rôle important dans le maintien de la stabilité génomique dans le stress oxydatif. KLF4 et ERK5 agissent ensemble pour protéger les neurones de l’apoptose induite par le stress oxydatif. Par conséquent, KLF4 peut agir comme une cible thérapeutique pour agir contre le stress oxydatif lorsqu’il est activé. Il a été rapporté que les statines peuvent activer ERK5, conduisant à l’expression de KLF4 et de ses gènes dépendants (Ohnesorge et al., 2010), mais le mécanisme reste flou, et les gènes cibles en amont et en aval liés à KLF4 sont moins étudiés dans le stress oxydatif, il est nécessaire de poursuivre l’étude.
Rôle de KLF4 dans la régénération des axones
La perte précoce des axones est une caractéristique commune des maladies neurodégénératives. La perte synaptique et la déficience du transport dans la MA peuvent entraîner des troubles cognitifs (Holtzman et al., 2011; Coleman, 2013). Le degré de dommages déclaratifs à la mémoire est lié à la densité synaptique de l’hippocampe et du cortex. Les oligomères Aß solubles réduisent l’absorption du glutamate et favorisent un dysfonctionnement synaptique, perturbant la plasticité synaptique (Li et al., 2009). Par conséquent, il est particulièrement important d’étudier comment réparer les axones dans le SNC. Dans les cellules ganglionnaires rétiniennes, les axones ont une forte capacité de croissance et de régénération au début du développement, mais dans le SNC des mammifères adultes, les axones perdent leur capacité de régénération et les neurones peuvent mourir ou s’atrophier (Goldberg et Barres, 2000; Goldberg et al., 2002).
KLF4 joue un rôle important dans l’inhibition de la croissance des axones. Dans les RGC embryonnaires, la surexpression de KLF4 peut réduire le pourcentage d’élongation des neurites, la longueur des axones et des dendrites et la ramification des neurites. En outre, il a été constaté que la surexpression de KLF4 peut réduire les taux de croissance des axones postnataux à long terme, mais n’a pas réussi à réduire les taux de croissance des axones à court terme (Moore et al., 2009; Steketee et coll., 2014). Des études ultérieures ont montré que les faisceaux d’axones des souris KLF4–cKO étaient plus épais que les souris témoins (Fang et al., 2016). De plus, l’élimination de l’expression de KLF4 pendant le développement peut augmenter le potentiel de reproduction des RGC adultes. De plus, KLF4 dépourvu du domaine de liaison à l’ADN c-terminal n’a eu aucun effet sur la croissance de l’axone. Il n’y a eu aucun impact sur la survie des cellules après la lésion des cellules ganglionnaires rétiniennes si le KLF4 était assommé (Moore et al., 2009).
KLF4 peut également affecter la régénération axonale. Une étude récente a rapporté que la diminution de l’expression de KLF4 dans les cellules ganglionnaires rétiniennes adultes favorisait la régénération des axones par la voie JAK-STAT3 (Qin et al., 2013). KLF4 a augmenté la phosphorylation de STAT3 et a régulé la croissance des axones via la signalisation JAK-STAT (Qin et Zhang, 2012). Sous le traitement des cytokines, les membres de la famille des protéines STAT sont phosphorylés aux sites carboxy-terminaux tyrosine et sérine à l’intérieur de la cellule pour former un dimère stable. Cette modification améliore la transcription des gènes associés aux cellules (Yuan et al., 2005). L’interaction entre KLF4 et STAT3 sur la phosphorylation induite par les cytokines de la tyrosine705 inhibe l’expression de STAT3 en inhibant la liaison de STAT3 à l’ADN (Qin et al., 2013). KLF4 knockdown améliore évidemment la régénération de l’axone dans les cellules ganglionnaires de la rétine après une lésion du nerf optique et empêche le nerf de se blesser après une légère lésion cérébrale. Les actions sont médiées par une diminution de p-p53 et une augmentation des niveaux de pSTAT3. KLF4 régule positivement l’apoptose neuronale via les voies p53 et JAK-STAT3, et KLF4 régule négativement la réparation axonale via la voie JAK-STAT3 (Cui et al., 2017).
Par conséquent, nous avons émis l’hypothèse que dans la MA, la régénération axonale peut être accomplie en modifiant l’expression de KLF4 ou en modifiant les voies de signalisation liées intracellulaires, et en contrôlant la progression de la MA en réduisant les axones manquants ou en réduisant le dysfonctionnement axonal. Cependant, la façon d’utiliser le facteur de transcription KLF4 dans des thérapeutiques potentielles doit encore être explorée.
Rôle de KLF4 dans l’accumulation de fer
Le fer est largement présent dans les systèmes biologiques, les métalloprotéinases liées au fer jouent un rôle clé dans le transport de l’oxygène, le transfert des électrons et la catalyse des réactions biochimiques (Aisen et al., 2001). Cependant, tout excès de fer au-delà de la plage physiologique normale peut nuire à la santé humaine (Adlard et Bush, 2006). Des études ont montré que la teneur en fer de l’hippocampe est corrélée négativement avec la performance des tests de mémoire (Ding et al., 2009). L’augmentation de la charge en fer dans le cerveau accélère la formation de plaques Aß et d’enchevêtrements de tau hyperphosphorylés, tout en augmentant le stress oxydatif (Peters et al., 2015). Le fer, qui a un degré élevé de perméabilité, favorise la croissance nerveuse et les connexions de cellule à cellule pendant le développement du cerveau (Dallman et Spirito, 1977).
Une étude récente a démontré que le stress physiologique provoquait l’activation de la voie de signalisation KLF4-HCP1 et une augmentation de l’absorption de l’hème (Li H. et al., 2017). L’hème représente 95% du fer fonctionnel dans le corps humain. C’est l’un des principaux composants de l’hème oxygénase (Hooda et al., 2014; Kurucz et coll., 2018). L’augmentation de l’activité de l’oxygénase-1 peut retarder l’oxydation du cerveau vieillissant (Verdile et al., 2015; Serini et Calviello, 2016; Kurucz et coll., 2018). Cela a un effet de soulagement sur AD. Le stress physiologique induit une augmentation du taux de glucocorticoïdes, le glucocorticoïde augmente l’expression de la protéine porteuse de l’hème 1 (HCP1) via KLF4, puis HCP1 favorise l’absorption de l’hème (Li H. et al., 2017). Les glucocorticoïdes et le KLF4 régulent ensemble les gènes anti-inflammatoires, et les cellules à faible teneur en glucocorticoïdes ne peuvent pas induire complètement l’expression du KLF4 (Sevilla et al., 2015). L’augmentation de la consommation d’hème induite par KLF4 entraîne une accumulation de fer dans le cerveau. Le fer favorise la libération de ROS (Tronel et al., 2013). L’élément fer améliore le stress oxydatif cérébral chez les rats soumis à un stress psychologique (Yu et al., 2011). Par conséquent, HCP1 peut être régulé par KLF4 et glucocorticoïde ensemble. L’augmentation du HCP1 améliore l’absorption de l’hème, ce qui entraîne directement une accumulation de fer dans le cerveau, exacerbe l’oxydation, augmente l’apoptose ou le dysfonctionnement et aggrave les lésions cérébrales.
Il est généralement admis que les troubles de la mémoire et de l’apprentissage sont les principaux symptômes de la MA. Un grand nombre de données cliniques ont montré que la charge de la plaque Aß et l’accumulation de fer réagissent au développement de l’apprentissage et du dysfonctionnement cognitif dans la MA (van Bergen et al., 2018). Des données récemment publiées suggèrent que le fer à forte dose augmente le dépôt d’Aß et atténue l’apprentissage et la mémoire chez la souris (Guo et al., 2013). Des études cliniques ont montré qu’une microglie contenant du fer se trouve dans l’hippocampe de patients atteints de MA sous imagerie par résonance magnétique (Zeineh et al., 2015). La microglie acquiert le fer à partir de sources extracellulaires et intracellulaires transférantes ou non transférantes (McCarthy et al., 2018). L’expression sélective et soutenue de KLF4 peut être induite dans le noyau et le cytoplasme des astrocytes réactifs de l’hippocampe ischémique (Park et al., 2014). Des études ont montré que KLF4 agit comme un répresseur transcriptionnel. Il régule l’expression de l’ELK-3, puis l’ELK-3 inhibe l’expression de HO-1 (Tsoyi et al., 2015). L’hème oxygénase-1 (HO-1) est une protéine de stress qui dégrade l’hème en bilirubine, en fer libre et en monoxyde de carbone. La régulation ascendante de HO-1 dans les astrocytes peut entraîner un dépôt anormal de fer et un dysfonctionnement mitochondrial dans le cerveau, entraînant une diminution des capacités cognitives (Schipper, 1999, 2004). Par conséquent, KLF4 peut être impliqué dans le processus d’accumulation de fer dans les astrocytes, exacerbant l’oxydation dans la MA et aggravant les lésions cérébrales.
Conclusion
KLF4 est communément connu pour jouer un rôle central dans la régulation de la prolifération cellulaire, de l’apoptose et de la différenciation. Des études antérieures se sont concentrées sur la régulation de KLF4 dans plusieurs processus neurophysiologiques importants, y compris la neuroinflammation, la neuroprotection et la régénération synaptique. Récemment, KLF4 s’est avéré jouer un rôle important dans la pathogenèse de la MA. Dans cet article, nous passons en revue le rôle de KLF4 dans la neuroprotection et la neurogenèse dans la MA.
KLF4 n’est pas seulement un régulateur de la régulation de la prolifération et de la différenciation cellulaires, mais aussi une cible potentielle pour réguler les réponses immunitaires. KLF4 peut réguler les facteurs inflammatoires négatifs et favoriser la réponse inflammatoire, et avoir un grand effet sur l’expression de la microglie nucléaire des astrocytes. De plus, KLF4 et ERK5 peuvent agir ensemble pour exercer des actions neuroprotectrices. De plus, la régénération des axones peut être réalisée en modifiant le contenu de facteurs de transcription spécifiques, d’inhibiteurs intracellulaires ou en modifiant les voies de signalisation intracellulaires. Éliminer KLF4 peut améliorer la régénération des axones et accélérer le taux de croissance des axones. La réduction de l’expression de KLF4 favorise la régénération des axones par la voie JAK-STAT3, et KLF4 favorise la voie JAK-STAT3 pour une régénération supplémentaire des axones. Par conséquent, KLF4 pourrait être impliqué dans le processus d’anti-inflammatoire, d’anti-apoptose, de régénération des axones et d’accumulation de fer dans le SNC, qui joue un rôle central dans la génération de la MA. Ces résultats suggèrent que KLF4 représente une cible thérapeutique potentielle pour la MA. Cependant, les mécanismes cellulaires et moléculaires profonds des effets de KLF4 sur la MA restent flous et des investigations supplémentaires sont nécessaires.
Contributions de l’auteur
ZQC, XHZ et YJ ont écrit le manuscrit. SHG et JYL ont modifié le cadre du manuscrit. BJL et RJC ont fourni les révisions critiques. Tous les auteurs ont approuvé la version finale du manuscrit pour soumission.
Financement
Ce travail a été soutenu par des subventions de la Fondation des Sciences naturelles de Chine (NSFC) (81871070, 31571126, 31471120) et du financement de l’Agence Jilin pour la Science et la Technologie (Subventions nos 20180519003JH, 20180414051GH, 20170414034GH et 20180414050GH).
Déclaration sur les conflits d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de relations commerciales ou financières pouvant être interprétées comme un conflit d’intérêts potentiel.
Cacabélos, R. (2018). Y a-t-il eu des améliorations dans la découverte de médicaments contre la maladie d’Alzheimer au cours des 5 dernières années? Exp. Opin. Drogue Discov. 13, 523–538. doi: 10.1080/17460441.2018.1457645
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Liu, Y., Shi, J. et Chen, X. (2015). Identification de nouvelles cibles pour la rhinite allergique saisonnière pendant et en dehors de la saison pollinique par analyse de microréseaux. Acta Oto-Rhino-Laryngol. 135, 1330–1336. doi: 10.3109 / 00016489.2015.1067906
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Serini, S., et Calviello, G. (2016). Réduction du stress oxydatif / nitrosatif dans le cerveau et son implication dans l’effet neuroprotecteur de l’AGPI n-3 dans la maladie d’Alzheimer. Curr. Alzheimer Res. 13, 123-134. doi: 10.2174/1567205012666150921101147
Résumé PubMed / Texte intégral croisé / Google Scholar