- Introduction
- Présentation du cas
- Matériaux et méthodes
- Caryotype et réseau SNP
- Séquençage de l’exome entier
- Extraction d’ARN, PCR et séquençage
- Résultats
- Discussion
- Disponibilité des données
- Déclaration d’éthique
- Contributions des auteurs
- Financement
- Déclaration de conflit d’intérêts
- Remerciements
- Matériel supplémentaire
Introduction
Le gène de la molécule d’adhésion cellulaire L1 (L1CAM) est une molécule d’adhésion cellulaire neuronale appartenant à la superfamille des immunoglobulines; il possède des fonctions clés dans le développement du système nerveux (Itoh et Fushiki, 2015). Les mutations de L1CAM ont été liées à des syndromes neurologiques liés à l’X, qui sont résumés comme des maladies L1. Ils sont classés comme suit: Hydrocéphalie liée à l’X (XLH) due à une sténose de l’aqueduc de Sylvius (HSAS), au syndrome de MASA (déficience intellectuelle, aphasie, démarche de brassage, pouces adduits), à une paraparésie spastique de type 1 (SP1) et à une agénésie liée à l’X du corps calleux (ACC) (Weller et Gartner, 2001; Itoh et Fushiki, 2015).
Environ 282 mutations pathogènes (DMs) dans le gène L1CAM ont été rapportées dans HGMD® Professional 2019.2 (https://portal.biobase-international.com/hgmd/pro/all.php). Les altérations du gène L1CAM sont variées; les analyses des données de mutation de 282 patients révèlent 51% de mutations erronées et non-sens, 25% de suppressions, 5% d’insertions et 19% de modifications du site d’épissure, mais les mutations silencieuses de L1CAM à potentiel pathogène étaient rares et les mutations silencieuses étaient souvent ignorées, en particulier lors de la détection du séquençage de l’exome entier (WES).
Dans cette étude, en utilisant WES, nous avons examiné l’ADN fœtal d’une femme enceinte chinoise qui a signalé cinq grossesses continues avec hydrocéphalie fœtale; nous n’avons trouvé qu’une nouvelle mutation silencieuse c. 453G > T (p. Gly151 =) dans le gène L1CAM. Fait intéressant, grâce à une analyse plus approfondie, nous avons indiqué que la mutation silencieuse créait une séquence consensuelle potentielle du site d’épissure 5 ‘, ce qui entraînerait une délétion dans la trame de 72 pb de l’exon 5 et de 24 acides aminés de la protéine L1CAM.
Présentation du cas
Une femme en bonne santé de 28 ans a été référée à notre clinique après quatre interruptions volontaires de grossesse dues à une hydrocéphalie fœtale dans d’autres hôpitaux. Tous les fœtus étaient des hommes. En arrivant à notre hôpital (Hôpital pour femmes, École de médecine, Université du Zhejiang, Zhejiang, Chine), elle en était déjà à sa cinquième grossesse à 24 semaines de gestation, avec une hydrocéphalie fœtale par examens d’image. Pour explorer la cause génétique, un prélèvement de sang fœtal a été effectué à 26 semaines d’âge gestationnel. Des études cytogénétiques conventionnelles ont été effectuées pour des échantillons fœtaux et parentaux, et l’échantillon fœtal a été analysé plus en détail par réseau de polymorphisme nucléotidique unique (SNP) et WES.
Cette étude a été réalisée conformément aux recommandations du Comité d’éthique de l’Hôpital pour femmes de l’École de médecine de l’Université de Zhejiang, et le consentement éclairé a été obtenu de tous les participants à cette étude conformément à la Déclaration d’Helsinki. Le protocole d’étude a été approuvé par le Conseil d’examen de l’Hôpital pour femmes de la Faculté de médecine de l’Université du Zhejiang en Chine.
Matériaux et méthodes
Caryotype et réseau SNP
Les caryotypes du sang de cordon fœtal et du sang de cordon périphérique ont été déterminés par caryotypage conventionnel d’au moins 30 lymphocytes sanguins, qui ont été arrêtés à la métaphase par les colchicines. Des caryotypes de bandes G de cellules cultivées ont été réalisés au niveau de la bande 320-400 avec une résolution d’environ 10 Mo. La matrice SNP a été réalisée par la matrice CytoScan ™ HD (Affymetrix, États-Unis) selon les instructions du fabricant, avec environ 2 600 000 marqueurs, dont 750 000 sondes SNP et 1 900 000 sondes sans polymorphisme pour une couverture complète du génome entier. Les données ont été analysées par le logiciel Chas (Chromosome Analysis Suite) (Affymetrix, Santa Clara, CA) basé sur l’assemblage GRCh37/hg19. Le seuil de déclaration du résultat du numéro de copie a été fixé à 500 ko avec un nombre de marqueurs ≥50 pour les gains et à 200 ko avec un nombre de marqueurs ≥50 pour les pertes.
Séquençage de l’exome entier
La majeure partie de WES a été fournie par l’Institut de génomique de Beijing. L’ADN génomique a été extrait par un kit sanguin DNeasy (Qiagen, CA) puis fragmenté par Covaris LE220 (Massachusetts, USA) pour générer une banque d’extrémité appariée (200-250 pb). Toutes les bibliothèques amplifiées ont été réalisées sur la plate-forme BGISEQ-500, l’ADN simple brin a été mélangé avec le kit de préparation de bibliothèque d’ADN MGIEasy ™ V1 (BGI, Shenzhen, Chine), puis séquencé en utilisant la chimie 100SR avec le kit de séquençage à haut débit BGISEQ-500RS (BGI, Shenzhen, Chine).
Des lectures propres (d’une longueur de 90 pb) dérivées du séquençage et du filtrage ciblés ont ensuite été alignées sur la référence du génome humain (hg19) à l’aide du progiciel Multi-Vision Burrows-Wheeler Aligner (BWA) (Li et Durbin, 2009). Après l’alignement, les fichiers de sortie ont été utilisés pour effectuer une couverture de séquençage et une analyse approfondie de la région cible, des variants à un nucléotide (SNVs) et des appels indel, nous avons utilisé le logiciel GATK pour détecter les SNVs et les indels (McKenna et al., 2010), tous les SNV et indels ont été filtrés et estimés via plusieurs bases de données, y compris la Base de données sur le polymorphisme mononucléotidique (dbSNP) du National Center for Biotechnology Information (NCBI), HapMap, l’ensemble de données du projet 1000 Genomes et la base de données de 100 adultes chinois en bonne santé. Nous avons utilisé Condel, SIFT, PolyPhen-2, LRT, Mutation Taster et PhyloP pour prédire l’effet des variantes. Les variants pathogènes sont évalués selon le protocole publié par l’American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015). La Base de données sur les mutations génétiques humaines (HGMD) a été utilisée pour dépister les mutations. Tous les variants pathogènes potentiels ont été validés à l’aide des méthodes de séquençage de Sanger.
Extraction d’ARN, PCR et séquençage
Les cellules mononucléées du sang périphérique (PMBC) et les cellules mononucléées du sang de cordon (CBMC) ont été isolées par séparation par gradient de densité Ficoll. L’ARN total a été extrait des PMBC et des CBMC à l’aide de TRIzol (Takara, Japon). Les ARN totaux extraits ont été transcrits en inverse à l’aide du kit RT (Takara, Japon). La PCR a été réalisée à l’aide de GoldStar Best Master Mix (CWBIO, Beijing). Les séquences d’amorces sont répertoriées : L1CAM-DNA-5F, CCCACCCGTCCTTTCCTA; L1CAM-DNA-5R, CGCTCGTCCTGCTTGATGT; L1CAM – ARNm-4-6- F, GGTGTCCACTTCAAACCCAA; et L1CAM-ARNm-4-6- R, GCGGCTTCTGTCAATCA. Le séquençage de Sanger a été effectué par un analyseur d’ADN ABI 3130.
Résultats
Une femme en bonne santé de 28 ans a été référée à notre clinique après quatre interruptions volontaires de grossesse dues à une hydrocéphalie fœtale. Tous les fœtus étaient des hommes (figure 1A). Le pedigree familial semblait montrer XLH. Elle était déjà en cinquième grossesse à 26 semaines de gestation. La ventriculomégalie fœtale a été détectée par échographie fœtale et IRM, qui ont systématiquement démontré la présence d’hydrocéphalie. Ils ont montré que le ventricule cérébral bilatéral et le troisième ventricule étaient évidemment dilatés, et qu’il y avait une hydrocéphalie sévère dans l’intracérébral et l’agénésie du corps calleux (Figure 1B).
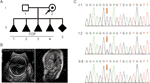
Figure 1 (A) Pedigree de la famille. HAUT, interruption de grossesse. B) Examens d’imagerie du fœtus. L’échographie fœtale et l’IRM fœtale ont montré qu’il y avait une hydrocéphalie sévère chez le fœtus. C) Analyse séquentielle de l’ADN génomique des membres de la famille. Les génotypes de L1CAM étaient de type sauvage, c. 453G > T Het, et c. 453G > T Hom, dans I:1 (mari), I: 2 (femme enceinte) et II: 5 (fœtus). La mutation est indiquée par les flèches rouges.
Afin d’explorer la cause génétique possible, nous avons effectué une analyse du caryotype et un réseau de SNP pour analyser le prélèvement de sang fœtal et n’avons trouvé aucun résultat positif. Le résultat de la néovascularisation choroïdienne (CNV) a été déposé dans l’Expression génique Omnibus (GEO) ; le numéro d’accession est GSE133063, tel qu’annexé ci-dessous (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
Ensuite, nous avons détecté le fœtus par WES. La stratégie analytique pour trouver une identification de variant pathogène probable est illustrée à la figure S1. Une liste de variantes (tableau S1) a été obtenue par le criblage des fréquences des variantes, de l’état de mutation et du mode d’héritage. En prenant en considération les gènes associés à l’hydrocéphalie (HP: 0000238, http://compbio.charite.de/hpoweb/showterm?id=HP:0000238#id=HP_0000238) (Tableau S2), il n’y a pas eu de mutation notable supplémentaire à l’exception de la mutation silencieuse de c.453G > T dans l’exon 5 du gène L1CAM (NM_000425.3). c. 453G > T n’a pas été signalé dans HGMD et ClinVar et n’a pas été trouvé dans dbSNP, gnomAD et d’autres ensembles de données. Selon les normes et directives de l’ACMG (Richards et al., 2015), il n’avait pas encore atteint le critère de « pathogène » ou « probablement pathogène », mais il n’y avait pas d’autres mutations potentielles; nous n’avions d’autre choix que de faire une analyse plus approfondie de la mutation silencieuse trouvée.
Selon la pensée traditionnelle, cette substitution de base s’est produite dans la troisième base du codon 151, qui code pour une glycine, créant ainsi une mutation neutre (p. Gly151 =). Cette variante a été confirmée dans l’ADN extrait du sang de cordon fœtal et du sang périphérique du couple par séquençage de Sanger (Figure 1C). La femme portait la mutation hétérozygote et son mari était un génotype de type sauvage.
Avec la mutation Taster (http://www.mutationtaster.org/), c. 453G > T a été noté comme « causant la maladie. »Cela a montré que les caractéristiques des protéines pouvaient être affectées et que le site d’épissure pouvait être modifié; nous étions curieux des effets d’épissage potentiels de la fonction L1CAM dans cette mutation silencieuse. La mutation silencieuse a été testée à l’aide des logiciels en ligne suivants: NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) et NNSplice (http://www.fruitfly.org/seq_tools/splice.html); le site d’épissure potentiel de 5′ a également été prédit pour être créé dans le c L1CAM.Mutation 453G >T à l’aide des produits logiciels (Figure S2). Les résultats ont montré que cette mutation silencieuse a créé un site d’épissure potentiel de 5′ à 72 pb en amont du site d’épissure normal de l’exon 6/intron 6 (Figure 2A). Si tel est le cas, nous pouvons trouver le changement de longueur de l’ARN messager L1CAM (ARNm) entre I:2 et II:5 (Figure 2A). La RT-PCR a été réalisée à l’aide d’amorces conçues pour amplifier les exons 4-6 dans l’ARNm L1CAM. En effet, les résultats ont montré une courte bande de troncature dans la PCR de l’ADNc fœtal (II: 5), tandis que la bande amplifiée à partir de l’ARNm L1CAM contenait la longue bande attendue dans la PCR de l’ADNc mari (I:1) et des bandes longues / courtes chez la femme enceinte ADNc PCR (I: 2) (Figure 2B). Le séquençage direct du fragment amplifié a montré que la délétion impliquait les 72 dernières pb de l’exon 5 dans l’ADNc fœtal mâle (la femme était porteuse) (Figure 2C). Nous avons les preuves pathogènes cruciales.
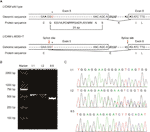
Figure 2 (A) Représentation schématique de l’organisation de l’exon 5, de l’intron 6 et de l’exon 6 dans L1CAM. (B) Analyse RT-PCR des exons 5 et 6 de l’ADNc L1CAM à partir de cellules mononucléées du sang périphérique (PMBC) et de cellules mononucléées du sang de cordon (CBMC). Électrophorèse sur gel d’agarose de produits RT-PCR générés à partir de I: 1 (mari), I: 2 (femme enceinte) et II: 5 (fœtus). (C) Analyse de séquence du produit RT-PCR à partir des PMBC du couple et des CBMC du fœtus.
Cette mutation silencieuse a entraîné 24 acides aminés de la protéine L1CAM (résidus 151-174) ; Lys(K) a été substitué par Glu(E) au codon 175 (Figure 2A). Il y a eu alignement de plusieurs séquences de protéines L1CAM chez plusieurs espèces et conservation des acides aminés manquants dans L1CAM chez les mammifères: Homo sapiens, Pan troglodytes, Bos taurus, Mus musculus et Rattus norvegicus (Figure 3A). Les protéines L1CAM de type sauvage et de mutation d’épissage c.453G > T ont été prédites par le logiciel serveur CPHmodels-3.2 (http://www.cbs.dtu.dk/services/CPHmodels/) (Figure 3B). Le domaine 2 de type immunoglobuline (de type Ig) (résidus 134-230) des protéines de type sauvage et de mutation d’épissage L1CAM est illustré à la figure 3C. La mutation d’épissage L1CAM c.453G > T a modifié la structure des protéines, en particulier le domaine de type Ig 2

Figure 3 (A) Alignement de plusieurs séquences de protéines L1CAM d’une espèce à l’autre. Le L1CAM c.453G > T ont entraîné l’absence de 24 acides aminés de la protéine L1CAM (résidus 151-174) dans la région des acides aminés conservés chez différentes espèces. La colonne noire montre les acides aminés manquants. (B) Les structures de la protéine L1CAM de type sauvage et de mutation d’épissage C.453G > T, telles que prédites par le serveur logiciel CPHmodels-3.2. (C) Les structures du domaine 2 de type immunoglobuline (type Ig) (résidus 134-230) de la protéine L1CAM de type sauvage et de mutation d’épissage.
Discussion
Des mutations silencieuses ont souvent été détectées par WES, mais une attention insuffisante a été accordée, ce qui a conduit à l’omission du DMs. Dans cette étude, nous avons utilisé WES pour explorer la cause génétique d’une famille chinoise atteinte d’hydrocéphalie, mais nous n’avons trouvé qu’une nouvelle mutation silencieuse dans L1CAM, ce qui nous a obligés à faire une analyse plus approfondie. Heureusement, nous avons prouvé que la mutation silencieuse a créé un nouveau site d’épissure de 5′ et était un DM.
Les mutations de L1CAM peuvent provoquer une maladie de L1 liée à l’X, mais les symptômes cliniques sont variables; les mutations produisent des phénotypes inattendus. Dans l’étude, les cinq fœtus souffrant sont tous des hommes, ce qui correspond à un schéma d’héritage. L’échographie fœtale et l’IRM montrent une maladie L1 typique, y compris la XLH et l’agénésie du corps calleux. Il améliore notre compréhension de la corrélation génotype-phénotype de L1CAM.
L1CAM c. 453G > T (p. Gly151 =) a initialement été pensé pour n’avoir aucun effet sur la séquence protéique. Mais d’autres mutations silencieuses, c. 924C > T (p. Gly308=) et c. 645C > T (p. Gly215 =), dans le gène L1CAM ont été rapportées comme étant des DMs (Du et al., 1998; Vos et coll., 2010). Le c.La mutation 924C > T a entraîné l’activation d’un nouveau site d’épissure de 69 pb 5′ au site d’épissure donneur normal de l’exon 8/intron 8, et il a été déclaré site » pathogène » pour l’hydrocéphalie (Du et al., 1998). Pour c.645C > T dans L1CAM, 51 pb ont été supprimés avec l’activation d’une nouvelle épissure donneuse exon 6/intron 6 (Vos et al., 2010). Notre étude actuelle était similaire; la mutation de c.453G > T a créé un site d’épissure potentiel de 5′ en amont du site d’épissure normal exon 5 /intron 5. Toutes ces mutations silencieuses ont créé de nouveaux sites d’épissure de donneurs, ce qui a entraîné l’abandon de l’exon. Il nous a rappelé de porter une attention particulière à ces mutations silencieuses, qui peuvent affecter l’épissage des protéines.
En tant que glycoprotéine transmembranaire et membre de la superfamille des molécules d’adhésion cellulaire des immunoglobulines, la protéine L1CAM peut interagir à la surface cellulaire avec un certain nombre de glycoprotéines différentes, et la liaison homophile est probablement son principal mode d’interaction (Wei et Ryu, 2012). Les études sur la structure cristalline des domaines de type Ig 1 à 4 dans la neurofascine ont suggéré que de nombreuses mutations pathologiques de la L1 affectent les résidus d’acides aminés conservés dans ces domaines et interfèrent avec les interactions homophiles (Liu et al., 2011), notamment tel que vérifié par la recherche de fonctions du domaine de type Ig 2 (Zhao et al., 1998). Dans notre étude, nous avons émis l’hypothèse que L1CAM c.453G > T a modifié le domaine de type Ig 2 dans la partie extracellulaire de la protéine L1CAM, conduisant à l’interaction extracellulaire anormale, sans commencer à initier la voie de signalisation en aval. Une autre étude consacrée à la spectrométrie de masse de cette variante L1CAM permettrait de clarifier spécifiquement quel ensemble moléculaire est produit dans la cellule.
En résumé, par l’intermédiaire de WES, nous avons signalé une nouvelle mutation silencieuse c. 453G > T dans L1CAM qui produit un site d’épissure de 5′ responsable de l’hydrocéphalie. On a prédit que cette variante protéique anormale modifierait le domaine de type Ig 2, ce qui pourrait affecter la liaison homophile de la protéine L1CAM. De plus, nous avons réalisé un diagnostic génétique prénatal pour la femme enceinte signalant cinq grossesses continues avec hydrocéphalie. Pendant ce temps, il a suggéré que certaines mutations silencieuses détectées chez WES ne devraient pas être ignorées; des prédictions d’épissage de ces mutations étaient nécessaires. Il a fourni une nouvelle base génétique pour le diagnostic prénatal et le diagnostic prénatal préimplantatoire de l’hydrocéphalie.
Disponibilité des données
Les ensembles de données accessibles au public ont été analysés dans le cadre de cette étude. Ces données peuvent être trouvées ici: GSE133063 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
Déclaration d’éthique
Les études impliquant des participants humains ont été examinées et approuvées par le Conseil d’examen de l’Hôpital pour femmes de la Faculté de médecine de l’Université du Zhejiang en Chine. Le consentement éclairé écrit pour participer à cette étude a été fourni par le tuteur légal / le plus proche parent des participants. Le consentement éclairé écrit a été obtenu de la ou des personnes, et du tuteur légal / proche parent du ou des mineurs, pour la publication de toute image ou donnée potentiellement identifiable incluse dans cet article.
Contributions des auteurs
YS, YLi, MC, YLu, YQ et YY ont mené des expériences. YS a préparé les chiffres. MC et YLi ont analysé les données de WES. YLu et YQ ont effectué une analyse du caryotype et un réseau de SNP. YY a recruté des échantillons. HL et FL ont fourni des examens d’imagerie. YS et MD ont écrit le manuscrit. Tous les auteurs ont lu et approuvé le manuscrit final.
Financement
Cette étude a été soutenue par la Fondation Nationale des Sciences Naturelles de Chine (Subvention No 81801441), le Programme clé de Recherche et Développement de la province du Zhejiang (Subvention No. 2019C03025), le Programme National de Recherche et Développement Clé de la Chine (Subvention n ° 2016YFC1000703) et la Fondation de la Recherche Scientifique Médicale de la province du Zhejiang (Subvention n ° 2014KYA246).
Déclaration de conflit d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de relations commerciales ou financières pouvant être interprétées comme un conflit d’intérêts potentiel.
Remerciements
Nous remercions les patients inscrits à cette recherche. Nous remercions le Dr. Jiong Gao (BGI Genomics, BGI-Shenzhen, Shenzhen 518083, Chine) pour son assistance lors de la préparation de ce manuscrit.
Matériel supplémentaire
Le matériel supplémentaire pour cet article peut être trouvé en ligne à l’adresse suivante:: https://www.frontiersin.org/articles/10.3389/fgene.2019.00817/full#supplementary-material
Figure S1 / Stratégie analytique pour trouver l’identification de variant pathogène probable par WES.
Figure S2 / Sites d’épissure donneurs prédits par NetGene2 et NNSplice.
Tableau S1 / Liste des variants grâce au criblage des fréquences des variants, de l’état de mutation et du mode d’héritage.
Tableau S2 / Liste des gènes associés à l’hydrocéphalie (Exportation pour HP: 0000238).
Du, Y. Z., Dickerson, C., Aylsworth, A. S., Schwartz, C.E. (1998). Une mutation silencieuse, C924T (G308G), dans le gène L1CAM entraîne une hydrocéphalie liée à l’X (HSAS). J. Med. Genet. 35 (6), 456–462. doi: 10.1136/jmg.35.6.456
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Itoh, K., Fushiki, S. (2015). Le rôle de L1cam dans la corticogenèse murine et la pathogenèse de l’hydrocéphalie. Pathol. Int. 65 (2), 58–66. doi : 10.1111/ pin.12245
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Li, H., Durbin, R. (2009). Alignement rapide et précis en lecture courte avec la transformation Burrows-Wheeler. Bioinformatique 25 (14), 1754-1760. doi: 10.1093/ bioinformatique / btp324
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Liu, H., Focia, P. J., He, X. (2011). Mécanisme d’adhésion homophile de la neurofascine, membre de la famille L1 des molécules d’adhésion des cellules neurales. J. Biol. Chem. 286 (1), 797–805. doi: 10.1074/ jbc.M110.180281
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Il s’agit de l’un des principaux facteurs de risque de la maladie d’alzheimer et de la maladie de Parkinson. (2010). La boîte à outils d’analyse du génome : un cadre MapReduce pour l’analyse des données de séquençage de l’ADN de nouvelle génération. Genome Res. 20 (9), 1297-1303. doi: 10.1101/gr.107524.110
Résumé PubMed / Texte intégral Croisé / Google Scholar
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi: 10.1038/ gim.2015.30
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Vos, Y. J., de Walle, H. E. K., Bos, K. K., Stegeman, J. A., Berge, A. M., Bruining, M., et al. (2010). Corrélations génotype-phénotype dans le syndrome L1: guide pour le conseil génétique et l’analyse des mutations. J.Med. Genet. 47 (3), 169–175. doi: 10.1136/jmg.2009.071688
Résumé PubMed / Texte intégral Croisé / Google Scholar
Wei, C. H., Ryu, S.E. (2012). Interaction homophile de la famille L1 des molécules d’adhésion cellulaire. Exp. Mol. Med. 44 (7), 413–423. doi: 10.3858/emm.2012.44.7.050
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Weller, S., Gartner, J. (2001). Aspects génétiques et cliniques de l’hydrocéphalie liée à l’X (maladie L1): mutations du gène L1CAM. Hum. Mutat. 18 (1), 1–12. doi: 10.1002/ humu.1144
PubMed Abstract | CrossRef Full Text | Google Scholar
Zhao, X., Yip, P. M., Siu, C. H. (1998). Identification of a homophilic binding site in immunoglobulin-like domain 2 of the cell adhesion molecule L1. J. Neurochem. 71 (3), 960–971 doi: 10.1046/j.1471-4159.1998.71030960.x.
PubMed Abstract | CrossRef Full Text | Google Scholar