La technologie de séquençage de l’ADN a été développée en 1977 grâce à Frederick Sanger. Il a fallu un peu plus de temps avant qu’il soit possible de séquencer un génome complet. En effet, nous avions besoin d’un modèle mathématique approprié et d’une puissance de calcul massive pour assembler des millions ou des milliards de petites lectures dans un génome complet plus grand. La puissance de calcul et les logiciels d’aujourd’hui sont la principale différence entre ce qui prenait des années de travail au début des années 2000 et ce qui ne prend que quelques heures aujourd’hui. L’algorithme que vous avez choisi pour ce faire est le « saint graal » de la technologie d’assemblage. Ces algorithmes intègrent l’une des variables les plus célèbres connues dans les modèles mathématiques, la k-mer.
L’origine du k-mer et du modèle mathématique qui l’entoure provient d’un mathématicien suisse de 1735, Leonhard Euler, connu comme le père de la fonction mathématique. Un mathématicien néerlandais Nicolaas de Bruijn a adapté les idées d’Euler pour trouver une séquence cyclique de lettres extraites d’un alphabet donné pour laquelle chaque mot possible d’une certaine longueur apparaît comme une chaîne de caractères consécutifs dans la séquence cyclique exactement une fois.
l’algorithme de de Bruijn a été adapté par des biologistes moléculaires, qui, de nombreuses années plus tard, ont été confrontés à un problème équivalent: comment assembler des séquences d’ADN. Ainsi, les scientifiques du monde entier utilisent maintenant le graphique De De Bruijn et la variable k.
Application de k-mers à l’assemblage de séquences d’ADN
En quelques mots, l’assemblage du génome de novo consiste à connecter de petites lectures d’ADN consécutives et à se retrouver avec des séquences plus grandes. Pour générer un graphe de de Bruijn (voir la figure ci-dessous), les nucléotides au bord de chaque lecture doivent chevaucher le bord d’un second (et ainsi de suite). L’objectif final est de créer un sommet consécutif, ce qui entraînera (potentiellement) de gros fragments d’ADN.
Vous devez fragmenter vos lectures en k-mers, qui sont un nombre spécifique de nucléotides qui se chevauchent. Le k-mer vous permet de générer une séquence unique à partir de nombreuses petites. Chaque séquence k-mer unique est identifiée et les copies supplémentaires sont éliminées. Cet aspect de k-mers vous permet de surmonter l’un des inconvénients du séquençage de nouvelle génération: obtenir des lectures qui représentent des régions génomiques avec des fréquences différentes (c’est—à-dire obtenir beaucoup de petites lectures d’une région). L’utilisation de k-mers élimine les séquences répétées plus d’une fois en raison d’une couverture de séquence inégale. Cependant, gardez à l’esprit qu’une faible taille de k-mer augmentera les chances de chevauchement des nucléotides, tandis qu’une valeur plus grande les diminuera.
La technologie d’assemblage de novo d’aujourd’hui est plus efficace lorsque vous utilisez des bibliothèques de grandes lectures (c’est–à-dire 1.000-10.000 bps) combinées à des bibliothèques plus petites (100-200 bps). Les logiciels peuvent utiliser la valeur k et k-mers pour assembler des lectures courtes. Ceux-ci peuvent ensuite être incorporés et vérifiés par des plus grands pour aboutir à des contigs plus précis.
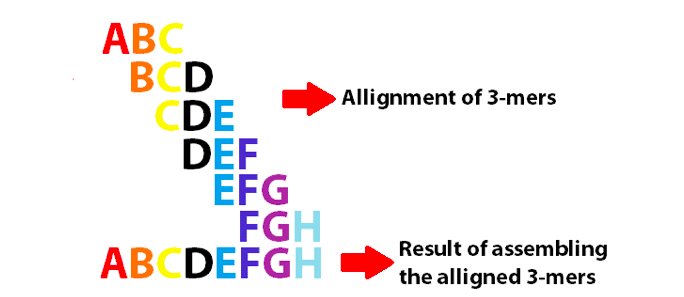
d’un graphe de de Bruijn utilisant 3-mers pour assembler les 8 premières lettres de l’alphabet anglais. Notez que ces 3-mers se chevauchent comme k-1.
Plus Vous En savez, plus Vous pouvez en obtenir dans l’assemblage de l’ADN
Il existe des conseils spécifiques à prendre en compte avant d’appliquer des graphiques De Bruijn dans votre méthode d’assemblage et de choisir la taille k-mer la plus appropriée. En les exploitant, vous pouvez générer de meilleurs résultats.
- Tout d’abord, et peut-être le plus important, est d’utiliser de nombreux k-mers différents dans votre assemblage. Vous devez ensuite évaluer vos résultats et choisir le (s) meilleur(s). N’oubliez jamais qu’il n’y a presque jamais un et un seul assemblage correct.
- Vous devez gérer soigneusement les lectures d’erreurs avant d’utiliser un k-mer. Si vous ne supprimez pas soigneusement les erreurs, les résultats peuvent créer un renflement indésirable, compliquant votre assemblage. Augmentez le seuil du taux d’erreur que vous utilisez lors du rognage de séquence. Vous risquez de perdre certaines séquences, mais celles qui restent seront les meilleures.
- Vous devez manipuler soigneusement les répétitions d’ADN. Par exemple, le séquençage Illumina génère une très grande quantité de données. Tout d’abord, essayez d’assembler une petite fraction des lectures, puis utilisez-les toutes pour repérer les différences. Des lectures courtes répétables peuvent interférer négativement avec votre processus d’assemblage.
- Connaissez vos données. Si vous ne connaissez pas la taille de votre génome attendu, la quantité de couverture de séquençage et le nombre de lectures, vous êtes plus enclin à choisir la meilleure valeur k pour assembler votre génome. Vous pouvez visiter les conseillers k-mer, comme velvet advisor de l’université Monash pour obtenir des conseils sur la valeur qui semble la plus appropriée.
L’utilisation de k-mers de différentes longueurs et l’alignement des contigs aident également les chercheurs à repérer les taux de mutation, élargissant ainsi son utilisation. Bien sûr, la manipulation des graphiques De De Bruijn vers l’avantage de l’assemblage n’est pas une panacée. Il y a beaucoup de choses à considérer qu’une fonction simpliste pour assembler le génome d’un organisme vivant. Ce n’est qu’une introduction à l’histoire et à la façon dont les biologistes peuvent l’utiliser plus efficacement.
- Compeau PE, Pevzner PA, Tesler G. (2011). Comment appliquer des graphiques de Bruijn à l’assemblage du génome.Biotechnologie de la nature. 29(11):987–91.
- Aggarwala V, Voight BF. (2016). Un modèle de contexte de séquence élargi explique largement la variabilité des niveaux de polymorphisme à travers le génome humain. Génétique de la nature. 48(4): 349–55.

Cela vous a-t-il aidé? Ensuite, veuillez partager avec votre réseau.