a DNS-szekvenálás technológiáját 1977-ben fejlesztették ki Frederick Sangernek köszönhetően. Egy kicsit tovább tartott, amíg sikerült egy teljes genomot szekvenálni. Ez azért van, mert megfelelő matematikai modellre és hatalmas számítási teljesítményre volt szükségünk ahhoz, hogy millió vagy milliárd apró olvasatot állítsunk össze egy nagyobb teljes genomba. A mai számítási teljesítmény és szoftver a fő különbség aközött, ami a 2000-es évek elején évekig tartott, és ami ma csak néhány órát vesz igénybe. Az algoritmus, amelyet erre választott, az összeszerelési technológia “Szent Grálja”. Ezek az algoritmusok magukban foglalják a matematikai modellekben ismert egyik leghíresebb változót, a k-mer-t.
az eredete a k-mer és a matematikai modell, amely körülveszi származik 1735 svájci matematikus Leonhard Euler, aki ismert, mint az apa a matematikai függvény. Nicolaas De Bruijn holland matematikus adaptálta Euler ötleteit, hogy megtalálja az adott ábécéből vett betűk ciklikus sorrendjét, amelyhez minden lehetséges, bizonyos hosszúságú szó pontosan egyszer jelenik meg egymást követő karakterek sorozataként a ciklikus szekvenciában.
de Bruijn algoritmusát molekuláris biológusok adaptálták, akik sok évvel később egyenértékű problémával szembesültek: hogyan állítsák össze a DNS-szekvenciákat. Így a tudósok világszerte a De Bruijn gráfot és a K változót használják.
a K-Mer-ek alkalmazása a DNS-szekvenciák összeállításához
néhány szóval a De novo genome assembly magában foglalja az egymást követő kis DNS-olvasások összekapcsolását és a nagyobb szekvenciák befejezését. A De Bruijn gráf létrehozásához (lásd az alábbi ábrát) az egyes leolvasások szélén lévő nukleotidoknak át kell fedniük a második szélét (stb.). A végső cél egy egymást követő csúcs létrehozása, amely (potenciálisan) nagy DNS-fragmenseket eredményez.
az olvasásokat k-Mer-ekbe kell bontani, amelyek egy meghatározott számú nukleotid, amelyek átfedik egymást. A k-mer lehetővé teszi, hogy egyedi szekvenciát hozzon létre sok kicsiből. Minden egyedi k-mer szekvenciát azonosítanak, és az extra másolatok megszűnnek. A k-mers ezen aspektusa lehetővé teszi a következő generációs szekvenálás egyik hátrányának leküzdését — olyan olvasások megszerzését, amelyek különböző frekvenciájú genomi régiókat képviselnek (vagyis sok kis olvasást kapnak egy régióból). A k-mers használata kiküszöböli a többször megismételt szekvenciákat az egyenlőtlen szekvenciafedettség miatt. Ne feledje azonban, hogy az alacsony k-mer méret növeli a nukleotidok átfedésének esélyét, míg a nagyobb érték csökkenti azokat.
a mai de novo összeszerelési technológia hatékonyabb, ha nagy olvasmányú (azaz 1000-10 000 bps) könyvtárakat használ kisebbekkel (100-200 bps) kombinálva. A szoftverprogramok a K értéket és a k-mers értéket használhatják a rövid olvasmányok összeállításához. Ezeket aztán beépíthetik és ellenőrizhetik a nagyobbak, hogy pontosabb összefüggésekbe kerüljenek.
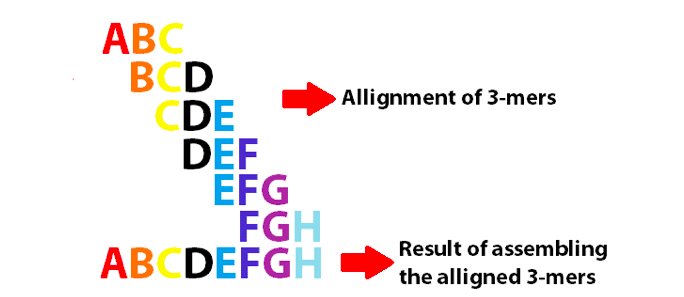
példa egy de Bruijn gráfra, amely 3 Mer-t használ az angol ábécé 8 első betűjének összeállításához. Vegye figyelembe, hogy ezek a 3 Mer-ek átfedik egymást k-1-ként.
minél többet tud, annál többet érhet el a DNS-összeállításban
vannak konkrét tippek, amelyeket figyelembe kell vennie, mielőtt de Bruijn grafikonokat alkalmazna az összeszerelési módszerben és kiválasztaná a legmegfelelőbb k-mer méretet. Ezek felhasználásával jobb eredményeket érhet el.
- először is, és talán a legfontosabb, hogy sok különböző k-Mer-t használj az összeállításban. Ezután értékelje az eredményeket, és válassza ki a legjobbat. Soha ne felejtsük el, hogy szinte soha nincs egyetlen helyes összeállítás.
- a K-mer használata előtt gondosan kezelje a hibaolvasásokat. Ha nem óvatosan távolítja el a hibákat, az eredmények nemkívánatos dudort hozhatnak létre, ami bonyolítja az összeszerelést. Növelje a sorozatvágás során használt hibaarány küszöbértékét. Lehet, hogy elveszít néhány szekvenciát, de azok, akik megmaradnak, a legjobbak lesznek.
- óvatosan kell kezelnie a DNS-ismétléseket. Például az Illumina szekvenálás nagyon nagy mennyiségű adatot generál. Először próbálja meg összeállítani az olvasások egy kis részét, majd használja őket a különbségek észlelésére. Az ismételhető rövid olvasmányok negatívan befolyásolhatják az összeszerelési folyamatot.
- Ismerje meg adatait. Ha nem ismeri a várható Genom méretét, a szekvenálási lefedettség mértékét és az olvasások számát, akkor hajlamosabb kiválasztani a legjobb k értéket a genom összeállításához. Látogasson el a k-mer tanácsadókhoz, mint például a Monash Egyetem bársony tanácsadója, hogy tanácsot kapjon arról, hogy melyik érték tűnik megfelelőbbnek.
a különböző hosszúságú k-Mer-ek használata és a kapcsolatok összehangolása szintén segíti a kutatókat a mutációk arányának észlelésében, kiterjesztve annak használatát. Természetesen a De Bruijn grafikonok manipulálása az összeszerelési előny felé nem csodaszer. Számos szempontot kell figyelembe venni, mint egy egyszerű funkciót az élő szervezet genomjának összeállításához. Ez csak egy bevezetés a történelem, és hogyan biológusok tudják használni hatékonyabban.
- Compeau PE, Pevzner PA, Tesler G. (2011). Hogyan kell alkalmazni a De Bruijn grafikonokat a genom összeállításához.Természet Biotechnológia. 29(11):987–91.
- Aggarwala V, Voight BF. (2016). A kibővített szekvencia-kontextus modell nagyjából megmagyarázza a polimorfizmus szintjének változékonyságát az emberi genomban. Természet Genetika. 48(4): 349–55.

segített ez neked? Akkor kérjük, ossza meg a hálózattal.