- Bevezetés
- eset bemutatása
- anyagok és módszerek
- kariotípus és SNP tömb
- teljes eXom szekvenálás
- RNS extrakciót, PCR-t és szekvenálást
- eredmények
- Vita
- adatok rendelkezésre állása
- etikai nyilatkozat
- szerzői hozzájárulások
- finanszírozás
- összeférhetetlenségi nyilatkozat
- Köszönetnyilvánítás
- Kiegészítő anyag
Bevezetés
az L1 sejtadhéziós molekula gén (L1CAM) az immunglobulin szupercsaládhoz tartozó neuronális sejtadhéziós molekula; kulcsfontosságú funkciókkal rendelkezik az idegrendszer fejlődésében (Itoh and Fushiki, 2015). Az L1CAM mutációi összefüggenek az X-hez kapcsolódó neurológiai szindrómákkal, amelyeket L1 betegségekként foglalnak össze. Ezek a következők szerint vannak besorolva: X-kapcsolt hydrocephalus (XLH) a Sylvius (HSAS) vízvezeték szűkületének, a MASA-szindrómának (értelmi fogyatékosság, afázia, csoszogó járás, adduktált hüvelykujj), az 1.típusú spasztikus paraparézisnek (SP1) és a corpus callosum (ACC) X-kapcsolt agenesisének (Weller and Gartner, 2001; Itoh and Fushiki, 2015) következtében.
körülbelül 282 betegséget okozó mutációt (DMS) jelentettek az L1CAM génben HGMD professional 2019.2 (https://portal.biobase-international.com/hgmd/pro/all.php). Az L1CAM gén változásai változatosak; a 282 betegből származó mutációs adatok elemzése 51% missense és nonszensz mutációt, 25% deléciót, 5% inszerciót és 19% splice helyváltoztatást tárt fel, de az l1cam patogén potenciállal rendelkező csendes mutációi ritkák voltak, és a csendes mutációkat gyakran figyelmen kívül hagyták, különösen a teljes exome szekvenálás (Wes) kimutatása során.
ebben a tanulmányban a WES segítségével átvilágítottuk egy kínai terhes nő magzati DNS-ét, aki öt folyamatos terhességről számolt be magzati hidrocefalussal; csak egy új néma mutációt találtunk c.453G > T (p.Gly151 = ) az L1CAM génben. Érdekes módon további elemzésekkel jeleztük, hogy a csendes mutáció potenciális 5′ splice site konszenzus szekvenciát hozott létre, ami 72 bp képkockán belüli delécióját eredményezné az l1cam fehérje 5.és 24. exon aminosavából.
eset bemutatása
egy 28 éves egészséges nőt utaltak klinikánkra négy önkéntes terhességmegszakítás után magzati hydrocephalus miatt más kórházakban. Minden magzat férfi volt. Amikor megérkezett a kórházunkba (Női Kórház, Orvostudományi Iskola, Zhejiang Egyetem, Zhejiang, Kína), már ötödik terhessége volt a terhesség 24.hetében, magzati hidrocefalussal képvizsgálatokkal. A genetikai ok feltárása érdekében a magzati vérmintát 26 hetes terhességi korban végezték. Hagyományos citogenetikai vizsgálatokat végeztek mind a magzati, mind a szülői mintákon, és a magzati mintát tovább elemezték egy nukleotid polimorfizmus (SNP) tömb és WES.
ezt a tanulmányt a Zhejiang Egyetem Orvostudományi Karának Etikai Bizottságának ajánlásaival összhangban végezték el, és a tanulmány minden résztvevőjétől tájékozott beleegyezést szereztek a Helsinki Nyilatkozatnak megfelelően. A vizsgálati protokollt a kínai Zhejiang Egyetem Női Kórházának Orvostudományi Karának felülvizsgálati testülete hagyta jóvá.
anyagok és módszerek
kariotípus és SNP tömb
a magzati köldökzsinórvér és a perifériás köldökzsinórvér kariotípusait legalább 30 vér limfocita hagyományos kariotipizálásával határozták meg, amelyeket metafázisban kolchicinek tartóztattak le. A tenyésztett sejtek G-sávos kariotípusait 320-400 sávos szinten végeztük, körülbelül 10 Mb felbontással. Az SNP tömböt a gyártó utasításai szerint a CytoScan ons HD array (Affymetrix, USA) végezte, mintegy 2 600 000 markerrel, köztük 750 000 SNP próbával és 1 900 000 nem polimorfizmus próbával a teljes genom átfogó lefedettsége érdekében. Az adatokat a kromoszóma analízis Suite (Chas) szoftver (Affymetrix, Santa Clara, CA) elemezte a GRCh37/hg19 összeállítás alapján. A másolatszám eredményének jelentési küszöbértékét 500 kb-ban határozták meg, nyereség esetén 50, veszteség esetén 200 kb-ban pedig 50 KB-ban.
teljes eXom szekvenálás
a Wes fő részét a pekingi genomikai Intézet biztosította. A genomi DNS-t egy DNeasy Vérkészlettel extraháltuk (Qiagen, CA), majd a Covaris le220 (Massachusetts, USA) fragmentálta, hogy párosított végű könyvtárat (200-250 bp) hozzon létre. Az összes amplifikált könyvtárat a BGISEQ-500 platformon hajtottuk végre, az egyszálú DNS-t összekeverjük MGIEasy adapterek DNS könyvtár Prep Kit V1 (BGI, Shenzhen, Kína), majd 100sr kémia segítségével szekvenáltuk BGISEQ-500Rs nagy áteresztőképességű szekvenáló készlettel (BGI, Shenzhen, Kína).
a célzott szekvenálásból és szűrésből származó tiszta leolvasásokat (90 bp hosszúsággal) ezután a burrows-Wheeler Aligner (BWA) Multi-Vision szoftvercsomag segítségével igazítottuk az emberi genom referenciához (hg19) (Li and Durbin, 2009). Az igazítás után a kimeneti fájlokat a célrégió szekvenálási lefedettségének és mélységének elemzésére, az egy nukleotid variánsokra (SNV) és az indel hívásokra használtuk, a GATK szoftvert használtuk az SNV-k és az Indel-ek kimutatására (McKenna et al., 2010), az összes SNV-t és Indel-t több adatbázison keresztül szűrték és becsülték meg, beleértve a Nemzeti Biotechnológiai Információs Központ (NCBI) egy nukleotid polimorfizmus adatbázisát (dbSNP), a HapMap-ot, az 1000 Genom Projekt adatkészletet és az 100 Kínai egészséges felnőtt adatbázisát. A Condel, a SIFT, a PolyPhen-2, az LRT, a Mutation Taster és a PhyloP segítségével megjósoltuk a variánsok hatását. A patogén variánsokat az American College of Medical Genetics and Genomics (ACMG) által kiadott protokoll alapján értékelik (Richards et al., 2015). A humán génmutációs adatbázist (HGMD) használták a mutációk szűrésére. Az összes lehetséges patogén variánst Sanger szekvenálási módszerekkel validáltuk.
RNS extrakciót, PCR-t és szekvenálást
perifériás vér mononukleáris sejteket (Pmbcs) és köldökzsinórvér mononukleáris sejteket (CBMCs) izoláltunk Ficoll sűrűséggradiens elválasztással. A teljes RNS-t extraháltuk a PMBCs-ből és a CBMCs-ből TRIzol (Takara, Japán) alkalmazásával. A kivont összes RNS-t reverz-átírtuk RT Kit (Takara, Japán) segítségével. A PCR-t GoldStar Best Master Mix (Cwbio, Peking) alkalmazásával végeztük. A Primer szekvenciák felsorolása: L1CAM-DNS-5F, CCCACCCGTCCTTTCCTA; L1CAM-DNS-5R, CGCTCGTCCTGCTTGATGT; L1CAM-mRNS-4-6-F, GGTGTCCACTTCAAACCCAA; és L1CAM-mRNS-4-6-R, GCGGCTTCCTGTCAATCA. A Sanger szekvenálást egy ABI 3130 DNS analizátor végezte.
eredmények
egy 28 éves egészséges nőt utaltak klinikánkra a terhesség négy önkéntes befejezése után a magzati hidrocefalusz miatt. Minden magzat hím volt (1a.ábra). Úgy tűnt, hogy a családi törzskönyv XLH-t mutat. Már itt volt ötödik terhesség nál nél 26 hetes terhesség. A magzati ventriculomegáliát magzati ultrahangvizsgálattal és MRI-vel detektálták, amely következetesen bizonyította a hydrocephalus jelenlétét. Kimutatták, hogy a bilaterális agykamra és a harmadik kamra nyilvánvalóan kitágult, és súlyos hydrocephalus volt a corpus callosum intracerebrális és agenesisében (1b ábra).
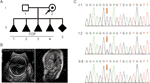
1. ábra (A) A család származása. TOP, a terhesség megszüntetése. B) a magzat képalkotó vizsgálata. A magzati ultrahang vizsgálat és a magzati MRI kimutatta, hogy súlyos hydrocephalus volt a magzatban. C) a családtagoktól származó genomi DNS Szekvenciaelemzése. Az L1CAM genotípusai vad típusúak voltak, c.453g > T Het és c. 453g > T Hom, I-ben:1 (férj), I:2 (terhes nő), II:5 (magzat). A mutációt a piros nyilak jelzik.
a lehetséges genetikai ok feltárása érdekében kariotípus elemzést és SNP tömböt végeztünk a magzati vérminták elemzésére, és nem találtunk pozitív eredményeket. A choroidalis neovaszkularizáció (CNV) eredménye lerakódott a génexpresszióban Omnibus (GEO); a csatlakozási szám az GSE133063, az alábbiak szerint (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
ezután Wes észlelte a magzatot. A valószínű patogén variáns azonosításának analitikai stratégiáját az S1 ábra mutatja. A variánsok listáját (S1 táblázat) a variáns frekvenciák, a mutációs státusz és az öröklődési mód szűrésével nyertük. Figyelembe véve a hydrocephalus-asszociált géneket (HP:0000238, http://compbio.charite.de/hpoweb/showterm?id=HP:0000238#id=HP_0000238) (S2 táblázat), nem volt további figyelemre méltó mutáció, kivéve a C.453g > T csendes mutációját az L1CAM gén 5.exonjában (NM_000425.3). c. 453G > T-T nem jelentettek HGMD-ben és ClinVar-ban, és nem találtak dbsnp-ben, gnomAD-ban és más adatkészletekben. Az ACMG szabványai és irányelvei szerint (Richards et al., 2015), még nem érte el a “patogén” vagy “valószínűleg patogén” kritériumot, de nem volt más lehetséges mutáció; nem volt más választásunk, mint a talált csendes mutáció további elemzése.
a hagyományos gondolkodás szerint ez a bázis szubsztitúció a 151 kodon harmadik bázisában történt, amely egy glicint kódol, ezáltal semleges mutációt hoz létre (P.Gly151 = ). Ezt a variánst a magzati köldökzsinórvérből és a perifériás vérből a párban Sanger szekvenálással kivont DNS-ben igazolták (1C ábra). A nő heterozigóta mutációt hordozott, férje vad típusú genotípus volt.
mutációs kóstolóval (http://www.mutationtaster.org/), c.453g > T “betegséget okozó.”Ez azt mutatta, hogy a fehérje jellemzői befolyásolhatók, és a splice helye megváltozhat; kíváncsiak voltunk az L1CAM funkció lehetséges splicing hatásaira ebben a néma mutációban. A néma mutációt a következő online szoftvertermékekkel tesztelték: NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) és NNSplice (http://www.fruitfly.org/seq_tools/splice.html); az 5′ potenciális splice helyet szintén az l1cam c-ben jósolták.453G > T mutáció a szoftvertermékek használatával (S2 ábra). Az eredmények azt mutatták, hogy ez a néma mutáció potenciális 5 ‘ 72 bp toldási helyet hozott létre a normál 6. exon/6. intron toldási helytől felfelé (2a. ábra). Ebben az esetben megtalálhatjuk az L1CAM messenger RNS (mRNS) hosszváltozását I:2 és II:5 között (2a ábra). Az RT-PCR-t a 4-6 exonok l1cam mRNS-ben történő amplifikálására tervezett primerek alkalmazásával végeztük. Valójában az eredmények rövid csonkolási sávot mutattak a magzati cDNS PCR – ben (II: 5), míg az l1cam mRNS-ből amplifikált sáv a férj cDNS PCR-ben (I:1) és hosszú/rövid sávok a terhes nő cDNS PCR (I:2) (2b ábra). Az amplifikált fragmens közvetlen szekvenálása azt mutatta, hogy a deléció magában foglalta az utolsó 72 bp exon 5-et a hím magzati cDNS-ben (a nő hordozó volt) (2C ábra). Megvan a létfontosságú kórokozó bizonyíték.
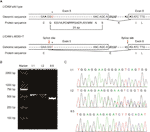
2. ábra (a) az 5., az intron 6. és a 6. exon szervezetének sematikus ábrázolása az L1CAM-ban. B) a perifériás vér mononukleáris sejtjeiből (pmbcs) és a köldökzsinórvér mononukleáris sejtjeiből (CBMCs) származó l1cam cDNS 5.és 6. exonjainak RT-PCR analízise. Az I:1 (férj), I:2 (terhes nő) és II:5 (magzat) RT-PCR termékek agarózgél-elektroforézise. C) A pár PMBCs-jéből és a magzat CBMCs-jéből származó RT-PCR termék Szekvenciaelemzése.
ez a néma mutáció 24 aminosav L1CAM fehérjét eredményezett (151-174 maradékok); A Lys (K) – t Glu (E) helyettesítette a 175 kodonon (2a.ábra). Több l1cam fehérje szekvenciát igazítottak több fajon keresztül, valamint az emlősökben az l1cam hiányzó aminosavainak megőrzését: Homo sapiens, Pan troglodytes, Bos taurus, Mus musculus és Rattus norvegicus (3a.ábra). Vad típusú és c.453G > t splicing mutáció az L1CAM fehérjéket a cphmodels-3.2 Server szoftver előre jelezte (http://www.cbs.dtu.dk/services/CPHmodels/) (3b ábra). Immunglobulin-szerű (Ig-szerű) domén 2 (maradékok 134-230) vad típusú és splicing mutáció L1CAM fehérjék ábrán látható 3C. L1CAM c. 453G > T splicing mutáció megváltoztatta a fehérje szerkezetét, különösen az Ig-szerű domén 2

3. ábra (A) több l1cam fehérjeszekvencia összehangolása Fajok között. Az L1CAM c.453G > T eredményeként 24 aminosav L1CAM fehérje (maradék 151-174) hiányzott a konzervált aminosav régióban különböző fajokban. A fekete oszlop a hiányzó aminosavakat mutatja. (B) a struktúrák vad típusú és c.453G > T splicing mutáció L1CAM fehérje által megjósolt szoftver CPHmodels-3.2 szerver. (C) a vad típusú és splicing mutáció l1cam fehérje immunglobulin-szerű (Ig-szerű) domén 2 (maradékok 134-230) szerkezete.
Vita
a Wes gyakran észlelt néma mutációkat, de nem fordítottak elegendő figyelmet, ami a DMs kihagyásához vezetett. Ebben a tanulmányban Wes-t alkalmaztuk egy hidrocefaluszos Kínai család genetikai okának feltárására, de csak egy új néma mutációt találtunk Az L1CAM-ben, ami további elemzésre kényszerített minket. Szerencsére bebizonyítottuk, hogy a néma mutáció egy új 5′ – es splice helyet hozott létre, és DM volt.
az L1CAM mutációi X-hez kapcsolódó L1 betegséget okozhatnak, de a klinikai tünetek változóak; a mutációk váratlan fenotípusokat eredményeznek. A tanulmányban az öt szenvedő magzat mind férfi, ami összhangban van az öröklési mintával. A magzati ultrahang vizsgálat és az MRI tipikus L1 betegséget mutat, beleértve az XLH-t és a corpus callosum agenesisét. Javítja az l1cam genotípus–fenotípus korrelációjának megértését.
L1CAM c.453G > T (p.Gly151 = ) eredetileg úgy gondolták, hogy nincs hatással a fehérje szekvencia. De más csendes mutációk, c.924C > T (p.Gly308 = ) és c.645c > T (p.Gly215 = ), az L1CAM génben DMs (Du et al., 1998; Vos et al., 2010). A c.A 924c > t mutáció egy új, 69 bp 5′ – es kötőhelyet aktivált a normál 8. exon / intron 8 donor kötőhelyre, és a hydrocephalus “betegséget okozó” helyének nyilvánították (Du et al., 1998). A C.645c > T esetében az L1CAM – ben az 51 bp-t egy új 6.exon/intron 6 donor toldás aktiválásával törölték (Vos et al., 2010). Jelen Vizsgálatunk hasonló volt; a C. 453g > T mutációja potenciális 5 ‘ illesztési helyet hozott létre a normál exon 5/intron 5 illesztési helyétől felfelé. Mindezek a néma mutációk új donor splice helyeket hoztak létre, ami az exon kihagyását eredményezte. Emlékeztetett minket arra, hogy fordítsunk nagy figyelmet ezekre a csendes mutációkra, amelyek befolyásolhatják a fehérjék splicingjét.
transzmembrán glikoproteinként és a sejtadhéziós molekulák immunglobulin szupercsaládjának tagjaként az L1CAM fehérje a sejtfelszínen számos különböző glikoproteinnel kölcsönhatásba léphet, és valószínűleg a homofil kötődés a kölcsönhatás fő módja (Wei and Ryu, 2012). A neurofascinben az Ig-szerű 1-4 domének kristályszerkezetével kapcsolatos vizsgálatok arra utaltak, hogy sok patológiás L1 mutáció befolyásolja a konzervált aminosavmaradékokat ezeken a doméneken belül, és zavarja a homofil kölcsönhatásokat (Liu et al., 2011), különösen az Ig-szerű 2.tartomány funkciókutatása alapján (Zhao et al., 1998). Vizsgálatunkban azt feltételeztük, hogy az L1CAM c. 453G > T megváltoztatta az Ig-szerű domént 2 az l1cam fehérje extracelluláris részében, ami abnormális extracelluláris interakcióhoz vezet, nem indul el a jelátviteli út lefelé történő kezdeményezése. Az L1CAM variáns tömegspektrometriájának szentelt további tanulmány pontosan tisztázza, hogy milyen molekuláris együttes keletkezik a sejtben.
összefoglalva, WES-en keresztül egy új néma mutációról számoltunk be C.453g > T az L1CAM-ben, amely egy 5′ – es illesztési helyet hoz létre, amely felelős a hydrocephalusért. Az előrejelzések szerint ez az abnormális fehérjeváltozat megváltoztatja az Ig-szerű domént 2, ami befolyásolhatja az L1CAM fehérje homofil kötődését. Ezenkívül prenatális genetikai diagnózist végeztünk a terhes nő számára, aki öt folyamatos terhességet jelentett hidrocefalusszal. Eközben azt javasolta, hogy a WES-ben észlelt néhány néma mutációt ne hagyja figyelmen kívül; ezeknek a mutációknak a splicing előrejelzéseire volt szükség. Új genetikai alapot biztosított a prenatális diagnózishoz és a hidrocefalusz prenatális diagnózisához.
adatok rendelkezésre állása
nyilvánosan elérhető adatkészleteket elemeztünk ebben a tanulmányban. Ezek az adatok itt találhatók: GSE133063 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
etikai nyilatkozat
az emberi résztvevőket érintő vizsgálatokat a kínai Zhejiang Egyetem Női Kórházának Orvostudományi testülete felülvizsgálta és jóváhagyta. Írásban tájékozott beleegyezés a tanulmányban való részvételhez a résztvevők törvényes gyámja/legközelebbi hozzátartozója. Az ebben a cikkben szereplő, potenciálisan azonosítható képek vagy adatok közzétételéhez az egyén(ek) től és a kiskorú(k) törvényes gyámjától/legközelebbi hozzátartozójától írásbeli tájékoztatáson alapuló beleegyezést kaptak.
szerzői hozzájárulások
Ys, YLi, MC, YLu, YQ és yy kísérleteket végzett. Ys elkészítette a számokat. MC és YLi elemezték a WES adatokat. Az YLu és az YQ kariotípus analízist és SNP tömböt végzett. Yy toborzott mintákat. A HL és az FL képalkotó vizsgálatokat végzett. YS és MD írta a kéziratot. Minden szerző elolvasta és jóváhagyta a végleges kéziratot.
finanszírozás
ezt a tanulmányt támogatta a National Natural Science Foundation of China (Grant No.81801441), a legfontosabb kutatási és fejlesztési Program a Zhejiang tartomány (Grant No. 2019c03025), Kína nemzeti kulcsfontosságú kutatási és Fejlesztési Programja (Grant No.2016yfc1000703), valamint Zhejiang tartomány Orvosi Tudományos Kutatási alapítványa (Grant No. 2014kya246).
összeférhetetlenségi nyilatkozat
a szerzők kijelentik, hogy a kutatást olyan kereskedelmi vagy pénzügyi kapcsolatok hiányában végezték, amelyek potenciális összeférhetetlenségnek tekinthetők.
Köszönetnyilvánítás
köszönjük a kutatásba bevont betegeket. Köszönjük Dr. Jiong Gao (BGI Genomics, BGI-Shenzhen, Shenzhen 518083, Kína) a kézirat elkészítése során nyújtott segítségért.
Kiegészítő anyag
a cikk kiegészítő anyaga megtalálható az interneten:: https://www.frontiersin.org/articles/10.3389/fgene.2019.00817/full#supplementary-material
S1 ábra / analitikai stratégia a valószínű patogén variáns Wes általi azonosításának megtalálásához.
S2 ábra / a NetGene2 és az NNSplice által előre jelzett Donorkötési helyek.
S1 táblázat / variánsok listája a variánsok gyakoriságának, mutációs állapotának és öröklődési módjának szűrésével.
S2 táblázat | a hydrocephalushoz kapcsolódó gének listája (Export HP:0000238).
Du, Y. Z., Dickerson, C., Aylsworth, A. S., Schwartz, C. E. (1998). Egy csendes mutáció, C924T (G308G), az L1CAM génben X-hez kapcsolt hydrocephalust (HSAS) eredményez. J. Med. Genet. 35 (6), 456–462. doi: 10.1136 / jmg.35.6.456
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Tudós
Itoh, K., Fushiki, S. (2015). Az L1cam szerepe az egér kortikogenezisében, valamint a hydrocephalus patogenezisében. Pathol. Int. 65 (2), 58–66. doi: 10.1111 / pin.12245
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Tudós
Li, H., Durbin, R. (2009). Gyors és pontos rövid olvasási igazítás a Burrows-Wheeler transzformációval. Bioinformatika 25 (14), 1754-1760. doi: 10.1093 / bioinformatika / btp324
PubMed absztrakt / CrossRef teljes szöveg / Google Scholar
Liu, H., Focia, P. J., ő, X. (2011). A neurofascin, az idegsejt-adhéziós molekulák L1 családjának tagja homofil adhéziós mechanizmusa. J. Biol. Kémia. 286 (1), 797–805. doi: 10.1074 / jbc.M110.180281
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
E., E., E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). A Genomelemzési eszközkészlet: MapReduce keretrendszer a következő generációs DNS-szekvenálási adatok elemzéséhez. Genom Res. 20 (9), 1297-1303. doi: 10.1101 / g.107524.110
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Szabványok és iránymutatások a szekvenciaváltozatok értelmezéséhez: az American College of Medical Genetics and Genomics és az Association for Molecular Pathology közös konszenzusos ajánlása. Genet. Med. 17 (5), 405–424. doi: 10.1038 / gim.2015.30
PubMed absztrakt / CrossRef teljes szöveg / Google Scholar
Vos, Y. J., de Walle, H. E. K., Bos, K. K., Stegeman, J. A., Berge, A. M., Bruining, M., et al. (2010). Genotípus-fenotípus korrelációk az L1 szindrómában: útmutató a genetikai tanácsadáshoz és a mutációelemzéshez. J. Med. Genet. 47 (3), 169–175. doi: 10.1136 / jmg.2009.071688
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Scholar
Wei, C. H., Ryu, S. E. (2012). A sejtadhéziós molekulák L1 családjának homofil kölcsönhatása. Felh. Mol. Med. 44 (7), 413–423. doi: 10.3858 / emm.2012.44.7.050
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Scholar
Weller, S., Gartner, J. (2001). Az X-kapcsolt hydrocephalus (L1 betegség) genetikai és klinikai vonatkozásai: az L1CAM gén mutációi. Hum. Mutat. 18 (1), 1–12. doi: 10.1002 / humu.1144
PubMed Abstract | CrossRef Full Text | Google Scholar
Zhao, X., Yip, P. M., Siu, C. H. (1998). Identification of a homophilic binding site in immunoglobulin-like domain 2 of the cell adhesion molecule L1. J. Neurochem. 71 (3), 960–971 doi: 10.1046/j.1471-4159.1998.71030960.x.
PubMed Abstract | CrossRef Full Text | Google Scholar