- Introduzione
- Presentazione del caso
- Materiali e metodi
- Cariotipo e Matrice SNP
- Sequenziamento dell’intero esoma
- Estrazione di RNA, PCR e sequenziamento
- Risultati
- Discussione
- Disponibilità dei dati
- Dichiarazione etica
- Contributi dell’autore
- Finanziamento
- Dichiarazione sul conflitto di interessi
- Riconoscimenti
- Materiale supplementare
Introduzione
L1 molecola di adesione delle cellule (gene L1CAM) è una molecola di adesione delle cellule neuronali appartiene alla superfamiglia delle immunoglobuline; possiede funzioni chiave nello sviluppo del sistema nervoso (Itoh e Fushiki, 2015). Le mutazioni in L1CAM sono state correlate a sindromi neurologiche legate all’X, che sono riassunte come malattie L1. Sono classificati come segue: Idrocefalo X-linked (XLH) a causa di stenosi dell’acquedotto di Sylvius (HSAS), sindrome di MASA (disabilità intellettiva, afasia, andatura mescolata, pollici addotti), paraparesi spastica di tipo 1 (SP1) e agenesia X-linked del corpo calloso (ACC) (Weller e Gartner, 2001; Itoh e Fushiki, 2015).
Circa 282 mutazioni che causano malattie (DMs) nel gene L1CAM sono state riportate in HGMD® Professional 2019.2 (https://portal.biobase-international.com/hgmd/pro/all.php). Le alterazioni nel gene L1CAM sono varie; le analisi dei dati sulle mutazioni di 282 pazienti rivelano il 51% di mutazioni missense e nonsense, il 25% di eliminazioni, il 5% di inserzioni e il 19% di cambiamenti del sito di giunzione, ma le mutazioni silenziose in L1CAM con potenziale patogeno erano rare e le mutazioni silenziose erano spesso ignorate soprattutto nel rilevamento del sequenziamento dell’intero esoma (WES).
In questo studio, utilizzando WES, abbiamo esaminato il DNA fetale di una donna incinta cinese che ha riportato cinque gravidanze continue con idrocefalo fetale; abbiamo trovato solo una nuova mutazione silenziosa c.453G > T (p.Gly151 = ) nel gene L1CAM. È interessante notare che, attraverso ulteriori analisi, abbiamo indicato che la mutazione silenziosa ha creato una potenziale sequenza di consenso del sito di giunzione 5′, che si tradurrebbe in una delezione in-frame di 72 bp da esone 5 e 24 aminoacidi della proteina L1CAM.
Presentazione del caso
Una donna sana di 28 anni è stata indirizzata alla nostra clinica dopo quattro interruzioni volontarie di gravidanza a causa di idrocefalo fetale in altri ospedali. Tutti i feti erano maschi. Quando arrivò al nostro ospedale (Ospedale femminile, Scuola di Medicina, Zhejiang University, Zhejiang, Cina), era già alla sua quinta gravidanza a 24 settimane di gestazione, con un idrocefalo fetale da esami di immagine. Per esplorare la causa genetica, il prelievo di sangue fetale è stato condotto a 26 settimane di età gestazionale. Sono stati eseguiti studi citogenetici convenzionali per campioni sia fetali che parentali e il campione fetale è stato ulteriormente analizzato mediante array di polimorfismo a singolo nucleotide (SNP) e WES.
Questo studio è stato condotto in conformità con le raccomandazioni del Comitato Etico del Women’s Hospital, School of Medicine Zhejiang University, e il consenso informato è stato acquisito da tutti i partecipanti a questo studio in conformità con la Dichiarazione di Helsinki. Il protocollo di studio è stato approvato dal Comitato di revisione dell’Ospedale femminile, School of Medicine, Zhejiang University in Cina.
Materiali e metodi
Cariotipo e Matrice SNP
I cariotipi del sangue del cordone fetale e del sangue del cordone periferico sono stati determinati mediante cariotipo convenzionale di almeno 30 linfociti del sangue, che sono stati arrestati a metafase dalle colchicine. I cariotipi G-banding delle cellule coltivate sono stati eseguiti a livello di banda 320-400 con una risoluzione di circa 10 Mb. L’array SNP è stato eseguito dall’array CytoScan ™ HD (Affymetrix, USA) secondo le istruzioni del produttore, con circa 2.600.000 marcatori, tra cui 750.000 sonde SNP e 1.900.000 sonde non polimorfiche per una copertura completa dell’intero genoma. I dati sono stati analizzati dal software Chromosome Analysis Suite (ChAS) (Affymetrix, Santa Clara, CA) basato sull’assemblaggio GRCh37/hg19. La soglia di segnalazione del risultato del numero di copia è stata fissata a 500 kb con un conteggio dei marcatori ≥50 per gli utili e a 200 kb con un conteggio dei marcatori ≥50 per le perdite.
Sequenziamento dell’intero esoma
La parte principale di WES è stata fornita dal Beijing Genomics Institute. Il DNA genomico è stato estratto da un kit di sangue DNeasy (Qiagen, CA) e poi è stato frammentato da Covaris LE220 (Massachusetts, USA) per generare una libreria accoppiata (200-250 bp). Tutte le librerie amplificate sono state eseguite sulla piattaforma BGISEQ-500, il DNA a singolo filamento è stato miscelato con MGIEasy™ DNA Library Prep Kit V1 (BGI, Shenzhen, Cina) e quindi sequenziato utilizzando la chimica 100SR con il kit di sequenziamento ad alto throughput BGISEQ-500RS (BGI, Shenzhen, Cina).
Le letture pulite (con una lunghezza di 90 bp) derivate dal sequenziamento e dal filtraggio mirati sono state quindi allineate al riferimento del genoma umano (hg19) utilizzando il pacchetto software Multi-Vision Burrows-Wheeler Aligner (BWA) (Li e Durbin, 2009). Dopo l’allineamento, i file di output sono stati utilizzati per eseguire la copertura sequenziamento e l’analisi di profondità della regione di destinazione, varianti a singolo nucleotide (SNVs), e indel chiamata, abbiamo usato il software GATK per rilevare SNVs e indel (McKenna et al., 2010), tutti gli SNV e gli indel sono stati filtrati e stimati tramite più database, tra cui il National Center for Biotechnology Information (NCBI) Single-Nucleotide Polymorphism Database (dbSNP), HapMap, 1000 Genomes Project dataset e database di 100 adulti sani cinesi. Abbiamo usato Condel, SIFT, PolyPhen-2, LRT, Mutation Taster e PhyloP per prevedere l’effetto delle varianti. Le varianti patogene sono valutate secondo il protocollo emesso dall’American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015). Il Human Gene Mutation Database (HGMD) è stato utilizzato per schermare le mutazioni. Tutte le potenziali varianti patogene sono state convalidate utilizzando metodi di sequenziamento Sanger.
Estrazione di RNA, PCR e sequenziamento
Le cellule mononucleate del sangue periferico (PMBCS) e le cellule mononucleate del sangue cordonale (CBMCs) sono state isolate dalla separazione del gradiente di densità di Ficoll. L’RNA totale è stato estratto da PMBCS e CBMCS usando TRIzol (Takara, Giappone). Gli RNA totali estratti sono stati trascritti al contrario utilizzando RT Kit (Takara, Giappone). La PCR è stata eseguita utilizzando GoldStar Best Master Mix (CWBIO, Pechino). Sono elencate le sequenze di primer: L1CAM-DNA-5F, CCCACCCGTCCTTTCCTA; L1CAM-DNA-5R, CGCTCGTCCTGCTTGATGT; L1CAM-mRNA-4-6-F, GGTGTCCACTTCAAACCCAA; e L1CAM-mRNA-4-6-R, GCGGCTTCCTGTCAATCA. Il sequenziamento Sanger è stato eseguito da un analizzatore di DNA ABI 3130.
Risultati
Una donna sana di 28 anni è stata indirizzata alla nostra clinica dopo quattro interruzioni volontarie di gravidanza a causa di idrocefalo fetale. Tutti i feti erano maschi (Figura 1A). Il pedigree familiare sembrava mostrare XLH. Era già qui quinta gravidanza a 26 settimane di gestazione. La ventricolomegalia fetale è stata rilevata mediante ecografia fetale e risonanza magnetica, che ha costantemente dimostrato la presenza di idrocefalo. Hanno mostrato che il ventricolo cerebrale bilaterale e il terzo ventricolo erano ovviamente dilatati, e c’era un grave idrocefalo nell’intracerebrale e nell’agenesia del corpo calloso (Figura 1B).
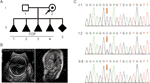
Figura 1 (A) Pedigree della famiglia. TOP, interruzione della gravidanza. (B) Esami di imaging del feto. L’ecografia fetale e la risonanza magnetica fetale hanno mostrato che c’era un grave idrocefalo nel feto. (C) Analisi di sequenza del DNA genomico dai membri della famiglia. I genotipi di L1CAM erano wild type, c. 453G > T Het, e c. 453G > T Hom, in I:1 (marito), I: 2 (donna incinta) e II: 5 (feto). La mutazione è indicata dalle frecce rosse.
Al fine di esplorare la possibile causa genetica, abbiamo eseguito l’analisi del cariotipo e l’array SNP per analizzare il prelievo di sangue fetale e non abbiamo trovato risultati positivi. Il risultato della neovascolarizzazione coroidale (CNV) è stato depositato nell’espressione genica Omnibus (GEO); il numero di adesione è GSE133063, come allegato di seguito (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
Quindi, abbiamo rilevato il feto da WES. La strategia analitica per trovare la probabile identificazione della variante patogena è stata mostrata nella figura S1. Un elenco di varianti (Tabella S1) è stato ottenuto attraverso lo screening delle frequenze delle varianti, dello stato delle mutazioni e della modalità di ereditarietà. Prendendo in considerazione i geni associati all’idrocefalo (HP:0000238, http://compbio.charite.de/hpoweb/showterm?id=HP:0000238#id=HP_0000238) (Tabella S2), non vi è stata alcuna mutazione notevole aggiuntiva ad eccezione della mutazione silenziosa di c.453G > T nell’esone 5 del gene L1CAM (NM_000425.3). c. 453G > T non è stato segnalato in HGMD e ClinVar e non è stato trovato in dbSNP, gnomAD e altri set di dati. Secondo gli standard e le linee guida dell’ACMG (Richards et al., 2015), non aveva ancora raggiunto il criterio di “patogeno” o “probabile patogeno”, ma non c’erano altre potenziali mutazioni; non avevamo altra scelta che fare un’ulteriore analisi della mutazione silenziosa trovata.
Secondo il pensiero tradizionale, questa sostituzione di base si è verificata nella terza base nel codone 151, che codifica una glicina, creando così una mutazione neutra (p.Gly151 = ). Questa variante è stata confermata nel DNA estratto dal sangue cordonale fetale e dal sangue periferico nella coppia mediante sequenziamento Sanger (Figura 1C). La donna portava la mutazione eterozigote e suo marito era un genotipo wild-type.
Con mutazione Assaggiatore (http://www.mutationtaster.org/), c.453G > T è stato segnato come “malattia causando.”Ha dimostrato che le caratteristiche proteiche potrebbero essere influenzate e il sito di giunzione potrebbe essere cambiato; eravamo curiosi dei potenziali effetti di splicing della funzione L1CAM in questa mutazione silenziosa. La mutazione silenziosa è stata testata utilizzando i seguenti prodotti software online: NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) e NNSplice (http://www.fruitfly.org/seq_tools/splice.html); il sito di giunzione potenziale 5′ è stato anche previsto per essere creato nel L1CAM c.453G > T mutazione utilizzando i prodotti software (Figura S2). I risultati hanno mostrato che questa mutazione silenziosa ha creato un potenziale sito di giuntura 5 ‘ 72 bp a monte del normale sito di giuntura esone 6/introne 6 (Figura 2A). Se questo è il caso, possiamo trovare la variazione di lunghezza dell’RNA messaggero L1CAM (mRNA) tra I:2 e II:5 (Figura 2A). La RT-PCR è stata eseguita utilizzando primer progettati per amplificare gli esoni 4-6 nell’mRNA L1CAM. Infatti, i risultati hanno mostrato una breve banda di troncamento in fetale cDNA PCR (II:5), mentre la banda amplificata da L1CAM mRNA conteneva la banda lunga atteso in marito cDNA PCR (I:1) e bande lunghe / corte nella donna incinta cDNA PCR (I: 2) (Figura 2B). Il sequenziamento diretto del frammento amplificato ha mostrato che la delezione ha coinvolto gli ultimi 72 bp di esone 5 nel cDNA fetale maschile (la donna era portatrice) (Figura 2C). Abbiamo le prove patogene cruciali.
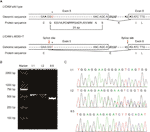
Figura 2 (A) Rappresentazione schematica dell’esone 5, dell’introne 6 e dell’esone 6 in L1CAM. B) Analisi RT-PCR degli esoni 5 e 6 del cDNA L1CAM da cellule mononucleate del sangue periferico (PMBCS) e cellule mononucleate del sangue cordonale (CBMCs). Elettroforesi su gel di agarosio di prodotti RT-PCR generati da I: 1 (marito), I:2 (donna incinta) e II:5 (feto). (C) Analisi di sequenza del prodotto RT-PCR da PMBCs della coppia e CBMCs del feto.
Questa mutazione silenziosa ha prodotto 24 aminoacidi della proteina L1CAM (residui 151-174); Lys (K) è stato sostituito da Glu (E) al codone 175 (Figura 2A). Ci sono stati l’allineamento di più sequenze proteiche L1CAM tra diverse specie e la conservazione degli amminoacidi mancanti in L1CAM tra i mammiferi: Homo sapiens, Pan troglodytes, Bos taurus, Mus musculus e Rattus norvegicus (Figura 3A). Le proteine wild-type e c. 453G > T splicing mutation L1CAM sono state previste dal software CPHmodels-3.2 Server (http://www.cbs.dtu.dk/services/CPHmodels/) (Figura 3B). Immunoglobulina-come (Ig-like) dominio 2 (residui 134-230) di wild-type e la mutazione di splicing L1CAM proteine è mostrato in Figura 3C. L1CAM c.453G > T mutazione di splicing alterato la struttura della proteina, in particolare l’Ig-like dominio 2

Figura 3 (A) Allineamento di più L1CAM sequenze di proteine in tutta la specie. Il L1CAM c.453G > T ha provocato 24 aminoacidi della proteina L1CAM (residui 151-174) mancanti nella regione amminoacidica conservata in diverse specie. La colonna nera mostra gli aminoacidi mancanti. (B) Le strutture di wild-type e c.453G > T splicing proteina mutazione L1CAM come previsto dal software CPHmodels-3.2 Server. (C) Le strutture del dominio 2 immunoglobulin-like (Ig-like) (residui 134-230) della proteina L1CAM di mutazione wild-type e splicing.
Discussione
Le mutazioni silenziose sono state spesso rilevate da WES, ma è stata prestata attenzione insufficiente, portando all’omissione di DMs. In questo studio, abbiamo impiegato WES per esplorare la causa genetica di una famiglia cinese con idrocefalo, ma abbiamo trovato solo una nuova mutazione silenziosa in L1CAM, che ci ha costretto a fare un’ulteriore analisi. Fortunatamente, abbiamo dimostrato che la mutazione silenziosa ha creato un nuovo sito di giunzione 5′ ed era un DM.
Le mutazioni in L1CAM possono causare una malattia L1 legata all’X, ma i sintomi clinici sono variabili; le mutazioni producono fenotipi inaspettati. Nello studio, i cinque feti sofferenti sono tutti maschi, il che è coerente con un modello ereditario. L’ecografia fetale e la risonanza magnetica mostrano una tipica malattia L1, tra cui XLH e agenesia del corpo calloso. Migliora la nostra comprensione sulla correlazione genotipo–fenotipo di L1CAM.
L1CAM c.453G > T (p.Gly151 = ) è stato inizialmente pensato per non avere alcun effetto sulla sequenza proteica. Ma altre mutazioni silenziose, c. 924C > T (p.Gly308 = ) e c.645C > T (p.Gly215 = ), nel gene L1CAM sono state segnalate come DMs (Du et al., 1998; Vos et al., 2010). Il c.La mutazione di 924C > T ha provocato l’attivazione di un nuovo sito della giuntura 69 bp 5 ‘al sito normale della giuntura del donatore dell’esone 8/intron 8 ed è stata dichiarata come sito” malattia-causante ” per l’idrocefalo (Du et al., 1998). Per c. 645C > T in L1CAM, 51 bp è stato eliminato con l’attivazione di una nuova giuntura del donatore esone 6 / introne 6 (Vos et al., 2010). Il nostro studio attuale era simile; la mutazione di c. 453G > T ha creato un potenziale sito di giuntura 5’ a monte del normale sito di giuntura esone 5/introne 5. Tutte queste mutazioni silenziose hanno creato nuovi siti di giuntura del donatore, con il risultato che l’esone viene saltato. Ci ha ricordato di prestare molta attenzione a queste mutazioni silenziose, che possono influenzare lo splicing delle proteine.
Come glicoproteina transmembrana e membro della superfamiglia delle immunoglobuline delle molecole di adesione cellulare, la proteina L1CAM può interagire sulla superficie cellulare con un numero di diverse glicoproteine e il legame omofilo è probabilmente la sua principale modalità di interazione (Wei e Ryu, 2012). Gli studi sulla struttura cristallina dei domini Ig-simili 1-4 nella neurofascina hanno suggerito che molte mutazioni patologiche di L1 influenzano i residui amminoacidici conservati all’interno di questi domini e interferiscono con le interazioni omofile (Liu et al., 2011), specialmente come verificato dalla ricerca sulla funzione del dominio Ig-like 2 (Zhao et al., 1998). Nel nostro studio, abbiamo ipotizzato che L1CAM c. 453G > T alterasse il dominio Ig-like 2 nella parte extracellulare della proteina L1CAM, portando all’interazione extracellulare anormale, non riuscendo a iniziare l’avvio a valle della via di segnalazione. Un ulteriore studio dedicato alla spettrometria di massa di questa variante L1CAM chiarirebbe specificamente quale insieme molecolare viene prodotto nella cellula.
In sintesi, attraverso WES, abbiamo riportato una nuova mutazione silenziosa c.453G > T in L1CAM che produce un sito di giunzione 5′ responsabile dell’idrocefalo. Si prevede che questa variante proteica anomala alteri il dominio Ig-like 2, che potrebbe influenzare il legame omofilo della proteina L1CAM. Inoltre, abbiamo eseguito la diagnosi genetica prenatale per la donna incinta riportando cinque gravidanze continue con idrocefalo. Nel frattempo, ha suggerito che alcune mutazioni silenziose rilevate in WES non dovrebbero essere ignorate; le previsioni di splicing di queste mutazioni erano necessarie. Ha fornito una nuova base genetica per la diagnosi prenatale e la diagnosi prenatale preimpianto di idrocefalo.
Disponibilità dei dati
I set di dati disponibili pubblicamente sono stati analizzati in questo studio. Questi dati possono essere trovati qui: GSE133063 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
Dichiarazione etica
Gli studi che hanno coinvolto partecipanti umani sono stati esaminati e approvati dal Comitato di revisione del Women’s Hospital, School of Medicine, Zhejiang University in Cina. Il consenso informato scritto a partecipare a questo studio è stato fornito dal tutore legale/parente più prossimo dei partecipanti. Il consenso informato scritto è stato ottenuto dall’individuo(s), e minore (s)’ tutore legale/parente più prossimo, per la pubblicazione di eventuali immagini o dati potenzialmente identificabili inclusi in questo articolo.
Contributi dell’autore
YS, YLi, MC, YLu, YQ e YY hanno condotto esperimenti. YS ha preparato le cifre. MC e YLi hanno analizzato i dati WES. YLu e YQ hanno eseguito analisi del cariotipo e array SNP. YY ha reclutato campioni. HL e FL hanno fornito esami di imaging. YS e MD hanno scritto il manoscritto. Tutti gli autori hanno letto e approvato il manoscritto finale.
Finanziamento
Questo studio è stato sostenuto dalla National Natural Science Foundation of China (Grant No. 81801441), il programma chiave di ricerca e sviluppo della provincia di Zhejiang (Grant no. 2019C03025), il programma nazionale di ricerca e sviluppo chiave della Cina (Grant No. 2016YFC1000703) e la Fondazione di ricerca scientifica medica della provincia di Zhejiang (Grant No. 2014KYA246).
Dichiarazione sul conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di relazioni commerciali o finanziarie che potrebbero essere interpretate come un potenziale conflitto di interessi.
Riconoscimenti
Ringraziamo i pazienti arruolati in questa ricerca. Ringraziamo il Dott. Jiong Gao (BGI Genomics, BGI-Shenzhen, Shenzhen 518083, Cina) per la sua assistenza durante la preparazione di questo manoscritto.
Materiale supplementare
Il materiale supplementare per questo articolo può essere trovato online all’indirizzo: https://www.frontiersin.org/articles/10.3389/fgene.2019.00817/full#supplementary-material
Figura S1 / Strategia analitica per trovare la probabile identificazione della variante patogena da parte di WES.
Figura S2 / Siti di giunzione donatori previsti da NetGene2 e NNSplice.
Tabella S1 / Elenco delle varianti attraverso lo screening delle frequenze delle varianti, dello stato delle mutazioni e della modalità di ereditarietà.
Tabella S2 / Elenco dei geni associati all’idrocefalo (Esportazione per HP:0000238).
Du, Y. Z., Dickerson, C., Aylsworth, AS, Schwartz, C. E. (1998). Una mutazione silenziosa, C924T (G308G), nel gene L1CAM provoca idrocefalo X-linked (HSAS). J. Med. Genet. 35 (6), 456–462. doi: 10.1136 / jmg.35.6.456
PubMed Abstract | CrossRef Testo completo / Google Scholar
Itoh, K., Fushiki, S. (2015). Il ruolo di L1cam nella corticogenesi murina e la patogenesi dell’idrocefalo. Pathol. Int. 65 (2), 58–66. doi: 10.1111 / perno.12245
PubMed Abstract / CrossRef Testo completo / Google Scholar
Li, H., Durbin, R. (2009). Veloce e preciso allineamento breve lettura con Burrows-Wheeler transform. Bioinformatica 25 (14), 1754-1760. doi: 10.1093 / bioinformatica / btp324
PubMed Abstract / CrossRef Full Text / Google Scholar
Liu, H., Focia, PJ, Lui, X. (2011). Meccanismo di adesione omofila della neurofascina, un membro della famiglia L1 delle molecole di adesione delle cellule neurali. J. Biol. Chimica. 286 (1), 797–805. doi: 10.1074 / jbc.M110.180281
PubMed Abstract / CrossRef Testo completo / Google Scholar
McKenna, A., Hanna, M., Banche, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). Il Genome Analysis Toolkit: un quadro MapReduce per l’analisi dei dati di sequenziamento del DNA di prossima generazione. Genome Res. 20 (9), 1297-1303. doi: 10.1101 / gr.107524.110
PubMed Abstract | CrossRef Testo completo / Google Scholar
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standard e linee guida per l’interpretazione delle varianti di sequenza: una raccomandazione di consenso congiunto dell’American College of Medical Genetics and Genomics e dell’Associazione per la patologia molecolare. Genet. Med. 17 (5), 405–424. doi: 10.1038 / gim.2015.30
PubMed Astratto | CrossRef Testo Completo | Google Scholar
Vos, Y. J., de Walle, H. E. K., Bos, K. K., Stegeman, J. A., Berge, A. M., Bruining, M., et al. (2010). Correlazioni genotipo-fenotipo nella sindrome L1: una guida per la consulenza genetica e l’analisi delle mutazioni. J. Med. Genet. 47 (3), 169–175. doi: 10.1136 / mgg.2009.071688
PubMed Abstract | CrossRef Testo completo / Google Scholar
Wei, CH, Ryu, S. E. (2012). Interazione omofila della famiglia L1 di molecole di adesione cellulare. Scad. Mol. Med. 44 (7), 413–423. doi: 10.3858 / emm.2012.44.7.050
PubMed Abstract | CrossRef Testo completo / Google Scholar
Weller, S., Gartner, J. (2001). Aspetti genetici e clinici dell’idrocefalo X-linked (malattia L1): mutazioni nel gene L1CAM. Ronzio. Mutato. 18 (1), 1–12. doi: 10.1002 / humu.1144
PubMed Abstract | CrossRef Full Text | Google Scholar
Zhao, X., Yip, P. M., Siu, C. H. (1998). Identification of a homophilic binding site in immunoglobulin-like domain 2 of the cell adhesion molecule L1. J. Neurochem. 71 (3), 960–971 doi: 10.1046/j.1471-4159.1998.71030960.x.
PubMed Abstract | CrossRef Full Text | Google Scholar