- Introduzione
- Le caratteristiche biologiche di KLF4
- Ruolo di KLF4 in AD
- Ruolo di KLF4 nella neuroinfiammazione
- Ruolo di KLF4 nell’apoptosi
- Ruolo del KLF4 nella rigenerazione degli assoni
- Ruolo di KLF4 nell’accumulo di ferro
- Conclusione
- Contributi dell’autore
- Finanziamento
- Dichiarazione sul conflitto di interessi
Introduzione
Il fattore 4 simile a Kruppel (KLF4) è un membro della famiglia del fattore di trascrizione del dito di zinco,che è espresso in vari tessuti umani. È ben noto come uno dei quattro fattori dell’induzione alle cellule staminali pluripotenti (iPSCs) (Ghaleb e Yang, 2017). KLF4 può regolare più importanti processi biologici come la neuroinfiammazione, lo stress ossidativo, la proliferazione, la differenziazione e l’apoptosi (Kaushik et al., 2010; Mamonkin et al., 2013; Zhang et al., 2015; Miao et al., 2017; Xu et al., 2017). Quantità di studi precedenti si sono concentrati sul ruolo di KLF4 nello sviluppo e nella progressione del cancro (Karam et al., 2017; Yadav et al., 2018). KLF4 è un fattore di trascrizione a doppia funzione, che può esercitare il suo ruolo come un oncogene o un gene soppressore del tumore a seconda del tipo di cancro o dello stadio del cancro (Evans e Liu, 2008). Può attivare o inibire la trascrizione di geni coinvolti nella proliferazione cellulare, differenziazione e apoptosi(Ding et al., 2015). KLF4 può collaborare con altri fattori di riprogrammazione per convertire le cellule somatiche in IPSC e inibire la differenziazione delle cellule staminali (Takahashi e Yamanaka, 2006; van Schaijik et al., 2018). Ciò fornisce prospettive terapeutiche per malattie vascolari, malattie immunitarie, anoressia e altre malattie (Imbernon et al., 2014; Liu Y. et al., 2015; Murgai et al., 2017). Inoltre, KLF4 può svolgere un ruolo ampiamente normativo nel sistema nervoso centrale (SNC). Diversi studi indicano che KLF4 è collegato a molteplici disturbi neurologici, tra cui il morbo di Alzheimer( AD), l’epilessia, il morbo di Parkinson, l’idrocefalo e la schizofrenia (Qin et al., 2011; Xie et al., 2013; Han et al., 2015; Nishiguchi et al., 2015; Li L. et al., 2017).
AD è una delle più comuni malattie neurodegenerative croniche, che porta a disturbi cognitivi e della memoria, vari sintomi mentali e anomalie comportamentali e demenza progressiva è la caratteristica clinica più comune (Jiang et al., 2018). Gli attuali fattori patogeni confermati di AD includono la formazione di placche senili indotte da deposizione anormale di amiloide-β (Aß) e i grovigli neurofibrillari o la neurite distrofica indotta dall’accumulo di tau (Querfurth e LaFerla, 2010; Shinohara et al., 2014). Inoltre, l’AD può essere influenzata anche da fattori genetici. Tuttavia, la patogenesi di suscitare è ancora oscura. I farmaci più diffusi per il trattamento dell’AD includono potenziatori di neurotrasmettitori, agenti anti-amiloidi, peptidi neuroprotettivi e altri farmaci (Cacabelos, 2018). In particolare, diversi studi hanno dimostrato che KLF4 ha svolto un ruolo significativo nella patogenesi dell’AD. In questa recensione, ci concentriamo sul ruolo regolatorio del KLF4 nella neuroinfiammazione, nell’apoptosi neuronale, nella rigenerazione assonale e nell’accumulo di ferro per spiegare l’associazione tra KLF4 e la patogenesi dell’AD, che potrebbe fornire approfondimenti sui meccanismi cellulari e molecolari dei disturbi neurodegenerativi.
Le caratteristiche biologiche di KLF4
KLF4 è una proteina nucleare contenente zinco, isolata dalla libreria NIH 3T3 e situata nel nucleo cellulare. E ” stato identificato e caratterizzato da Shields et al. (1996). La massa molecolare di KLF4 umano è 55kD e si trova sul cromosoma 9q31. KLF4 copre un segmento genico di 6,3 kb e ha cinque esoni. La sua regione codificante cDNA codifica un polipeptide costituito da 470 residui di aminoacidi (ancora et al., 1998; Ghaleb e Yang, 2017). Il terminale carbossilico di KLF4 ha una regione della struttura legante del DNA che contiene tre strutture del dito dello zinco del tipo Cys2His2 (C2H2), che sono formate da 81 amminoacidi altamente conservati. Regola la trascrizione per alta affinità con gli elementi CACCC e le sequenze di DNA del gene bersaglio ricco di GC (Shields e Yang, 1998; Pearson et al., 2008). La maggior parte dei siti di legame del DNA di KLF4 si trovano all’interno della regione del dito di zinco, incluso il dominio di attivazione della trascrizione N-terminale per l’interazione delle proteine, la struttura del dito di zinco C-terminale per il legame del DNA e la zona di inibizione della trascrizione (Bieker, 2001). KLF4 è coinvolto nella regolazione dell’espressione di molti geni endogeni (Shields e Yang, 1998). C’è un dominio regolatorio trascrizionale altamente variabile al termine amminico di KLF4. I residui amminoacidici situati tra il 91 e il 117 amminico costituiscono un dominio di attivazione trascrizionale, che è ricco di prolina e serina, mentre esiste anche un dominio di repressione trascrizionale. Pertanto, KLF4 ha due effetti avversi: attivazione e inibizione della trascrizione genica (Yet et al., 1998; Wei et al., 2006).
Durante lo sviluppo embrionale, KLF4 era più alto espresso nella fase avanzata dello sviluppo embrionale. Mentre nei tessuti e negli organi maturi, KLF4 è espresso principalmente nel tratto gastrointestinale, nella cavità orale, nell’epidermide della pelle, nell’endotelio vascolare e nel rene, ed è meno espresso nel cervello (Segre et al., 1999; Ghaleb et al., 2011; Liu et al., 2013; Chen et al., 2015; Lui et al., 2015; Bin et al., 2016). Si pensa che svolga un ruolo significativo nella regolazione della proliferazione e della differenziazione cellulare. Inoltre, KLF4 può anche regolare il ciclo cellulare. KLF4 può attivare P21 in modo P53-dipendente (Zhang et al., 2000). Inoltre, è stato riscontrato che le cellule KLF4 (–/–) sono entrate in fase di senescenza prima delle cellule KLF4 ( + / + ), il che può essere spiegato dall’espressione genica meno antiossidante e dal livello ROS (Reactive oxygen species) più elevato nelle cellule KLF4 ( – / – ). Il ROS può aumentare l’espressione di p53 e p21 e successivamente promuovere il danno al DNA (Liu C. et al., 2015). È stato trovato che PRMT5 può elevare l’espressione di KLF4 nei livelli della proteina. PRMT5 è stato segnalato per aumentare la trascrizione di p21 e diminuire l’espressione di bax via inibendo KLF4 ubiquitilazione (Hu et al., 2015). Inoltre, numerosi studi hanno dimostrato che KLF4 è coinvolto nella regolazione dell’apoptosi dei neuroni (Kong et al., 2016; Cui et al., 2017; Canzone et al., 2018). Ruolo regolatorio fisiologico di KLF4 che abbiamo conosciuto sono ancora poco e sono necessarie ulteriori indagini.
Ruolo di KLF4 in AD
È ben noto che AD è principalmente caratterizzato da disturbi della memoria e cognitivi e disfunzione esecutiva (Goedert e Spillantini, 2006). Molti studi hanno dimostrato che l’apoptosi neuronale e la disfunzione sinaptica sono basi patologiche del declino della funzione cognitiva (Caccamo et al., 2017; Guo et al., 2017; Yoon et al., 2018). Il danno accumulato della deposizione di Aß, dello stress ossidativo e dell’accumulo di ferro può portare a disfunzione neuronale e apoptosi dei pazienti con AD. Diversi studi hanno dimostrato che il ruolo normativo di KLF4 sembra essere cruciale nel SNC. Considerando che KLF4 è stato segnalato per regolare l’apoptosi neuronale, la rigenerazione sinaptica, lo stress ossidativo e la neuroinfiammazione, la relazione tra KLF4 e la patogenesi dell’AD potrebbe essere un potenziale nuovo bersaglio per il trattamento dell’AD.
Ruolo di KLF4 nella neuroinfiammazione
Quantità di studi clinici hanno dimostrato che Aß può aggregarsi ed è il componente principale dei depositi extracellulari del tessuto cerebrale dei pazienti con AD, che può compromettere le sinapsi e i neuroni circostanti e portare alla morte neuronale. La secrezione anormale o l’eccessiva produzione di Aß porta a cambiamenti patologici dell’AD, quindi la deposizione di Aß è il collegamento principale dell’AD (Rajmohan e Reddy, 2017). Inoltre, gli studi hanno dimostrato che un’eccessiva deposizione di Aß può stimolare le cellule gliali a secernere ROS e altri fattori influenzanti, portando a stress ossidativo. Era noto che lo stress ossidativo può stimolare la produzione di Aß. Pertanto, Aß e lo stress ossidativo possono interagire tra loro e influenzare la progressione dell’AD (Cheignon et al., 2018).
KLF4 è stato segnalato come un potenziale modulatore e ha un grande effetto sull’infiammazione mediando macrofagi e cellule endoteliali (Figura 1) (Yoshida et al., 2014; Kapoor et al., 2015; Yang et al., 2018). Nel SNC, reazioni infiammatorie eccessive e croniche possono causare danni ai neuroni e ai neurogliociti. Recentemente è stato dimostrato che l’espressione di KLF4 è correlata positivamente con la neuroinfiammazione indotta da Aß42. Nelle cellule microgliali BV2, l’oligomerico Aß42 può aumentare l’espressione di KLF4, che è mediata da P53 attivato (Li L. et al., 2017). In condizioni infiammatorie, come l’accumulo di Aß, il rilascio di citochine pro-infiammatorie può essere stimolato nella generazione di AD (Griffin e Barger, 2010). La potenza neurotossica e la potenza pro-infiammatoria degli oligomeri solubili Aß42 sono relativamente superiori al deposito di fibre insolubili (Selkoe, 1991; Weinberg et al., 2018). Il silenzio di KLF4 è in grado di ripristinare la neuroinfiammazione mediata da Aß42 e la sovraespressione di KLF4 può esacerbare la neuroinfiammazione mediata da Aß42 (Li L. et al., 2017). L’accumulo di Aß induce l’attivazione di astrociti e microglia (Rodríguez et al., 2016). Gli astrociti attivati possono migliorare la neuroinfiammazione rilasciando fattori pro-infiammatori come IL-1, IL-6 e TNF-α (Rubio-Perez e Morillas-Ruiz, 2012; Doméné et al., 2016). Il circolo vizioso delle risposte infiammatorie alla fine porta alla disfunzione e all’apoptosi neuronale.
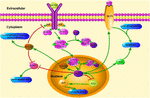
FIGURA 1. Schematicillustration delle vie di segnalazione relative a KLF4. Questa figura evidenzia il ruolo di KLF4 nella neuroprotezione e nella rigenerazione degli assoni. Le frecce nella figura indicano l’attivazione o la promozione e le linee rette indicano l’inibizione correlata. KLF4, fattore simile a Kruppel 4; STAT3, trasduttore di segnale e attivatore della trascrizione 3; JAK, Janus Chinasi; SOCS3, Soppressore della segnalazione delle citochine 3; HCP1, proteina portante dell’eme 1; ERK5, proteina chinasi attivata dal mitogeno 5.
KLF4 svolge un ruolo cruciale nella regolazione dei segnali pro-infiammatori. Nelle cellule gliali, l’attivazione KLF4 indotta da gemfibrozil aumenta il soppressore del segnale citochinico 3 (SOCS3) tramite la via PI3-chinasi-AKT (Ghosh e Pahan, 2012). Il knockdown mediato da siRNA di KLF4 potrebbe attenuare il livello dei SOC nell’astroglia e nella microglia dei topi, che potrebbero successivamente influenzare l’espressione del gene infiammatorio (Kaushik et al., 2010; Ghosh e Pahan, 2012). Inoltre, la cancellazione dei SOC può promuovere la sopravvivenza dei neuroni feriti e promuovere la rigenerazione degli assoni (Smith et al., 2009; Sun et al., 2011). E KLF4 regola positivamente la produzione di IL-1β o altri marcatori pro-infiammatori. Regola positivamente la cicloossigenasi – 2 (Cox-2) e regola negativamente l’inducibile ossido nitrico sintasi (iNOS) (Kaushik et al., 2013). Inoltre, KLF4 è un importante fattore regolatore per la differenziazione dei monociti e un potenziale bersaglio per la regolazione immunitaria (Alder et al., 2008). Pertanto, KLF4 potrebbe promuovere la neuroinfiammazione regolando questi regolatori negativi.
Vale la pena ricordare che nel modello di malattia di Parkinson, KLF4 può promuovere lo stress ossidativo indotto da MPP + e la neurotossicità, quindi aumentare l’apoptosi neuronale e ritardare la proliferazione cellulare (Chen et al., 2013). Lo stress ossidativo è uno squilibrio tra perossidazione e antiossidazione. I radicali liberi possono causare cambiamenti in diverse macromolecole, portando a danni cellulari, invecchiamento cellulare e danni ai tessuti (Parajuli et al., 2013; Nie et al., 2015). Lo stress ossidativo può aggravare l’infiammazione precoce e la produzione di Aß e quindi aggravare l’AD (Cai et al., 2011). Pertanto, KLF4 può essere coinvolto nello stress ossidativo nell’AD.
Questi risultati implicano KLF4 un ruolo chiave nel mediare la neuroinfiammazione attivando la microglia e il conseguente rilascio di citochine proinfiammatorie. Ha il potenziale per migliorare la neuroinfiammazione. Finora, molti studi sulla patogenesi dell’AD si sono concentrati sulla neuroinfiammazione. Come potenziale bersaglio per la regolazione immunitaria, KLF4 può promuovere le risposte infiammatorie della microglia attraverso l’influenza dei regolatori negativi correlati, che ha un grande effetto sullo sviluppo dell’AD.
Ruolo di KLF4 nell’apoptosi
I cambiamenti neurodegenerativi includono la graduale perdita di neuroni e sinapsi nelle regioni cerebrali rappresentative, come la corteccia cerebrale, l’ippocampo e altre regioni sottocorticali. Le menomazioni funzionali del SNC indotte dalla perdita neuronale sono permanenti (Citron, 2010). Lo stress ossidativo prolungato può portare all’apoptosi neuronale (Wu et al., 2010). Un gran numero di studi ha confermato che l’AD è strettamente correlata allo stress ossidativo (Lee et al., 2012; Yui et al., 2015). Si è constatato che lo stress ossidativo cronico può migliorare l’espressione della fosfolipasi A2 gruppo 3 (Pla2g3) in astrociti e interrompere l’equilibrio di Aß, e di conseguenza portare allo sviluppo di AD (Yui et al., 2015).
Molti studi hanno dimostrato che KLF4 svolge un ruolo importante nell’inibire lo sviluppo dello stress ossidativo (Shi et al., 2014; Liu C. et al., 2015). È stato trovato che KLF4 può promuovere l’apoptosi delle cellule indotta da H2O2, questa azione è probabilmente causata dall’aumento dell’espressione di bax e dalla diminuzione dell’espressione di bcl-2 (Li et al., 2010). La quercetina potrebbe ridurre l’espressione di KLF4 nelle cellule SH-SY5Y del neuroblastoma umano e aumentare l’espressione del rapporto bcl-2/bax. Inoltre, la quercetina può moderare il tasso di apoptosi della cellula SH-5YSY e ridurre l’attività dell’enzima caspasi-3 (Xi et al., 2012). Un recente studio ha studiato l’effetto neuroprotettivo della proteina attivata dal mitogeno (MAP) chinasi 5 (ERK5) contro lo stress ossidativo. L’attivazione di ERK5 può ridurre parzialmente la morte dei neuroni ippocampali indotti da H2O2 e aumentare la neuroprotezione indotta da NGF e PC(Su et al., 2014). Nils et al. utilizzato un mutante di MEK5 (MEK5D) per studiare la trascrizione attivata da ERK5 e le risposte funzionali nelle cellule endoteliali umane, e identificato KLF4 era un nuovo bersaglio ERK5 a valle (Ohnesorge et al., 2010). È stato riscontrato che la sovraespressione di KLF4 può sopprimere le risposte infiammatorie mediate dal TNF e ridurre l’adesione dei leucociti e l’apoptosi delle cellule basali. Questi risultati confermano che KLF4 ha proprietà antinfiammatorie e anti-apoptotiche (Ohnesorge et al., 2010). Esperimenti successivi hanno dimostrato che la scomparsa della malformazione cavernosa cerebrale 1 (CCM1) nelle cellule endoteliali attiva ERK5 tramite la via del segnale MEKK3-MEK5 e aumenta l’espressione di KLF4 (Cuttano et al., 2016). ERK5 svolge un ruolo di mediazione nel precondizionamento (PC) e nel fattore di crescita nervosa (NGF) up-regulated l’espressione di KLF4 (Su et al., 2014). Inoltre, l’abbattimento mediato da RNAi di KLF4 può anche ridurre la neuroprotezione indotta da NGF o PC. La sovraespressione di KLF4 porta a un più alto rapporto bcl-2/bax nelle cellule stressate da H2O2 (Su et al., 2014). KLF4 sovraespresso accelera i cambiamenti in bcl – 2 e bax combinando con il suo promotore corrispondente (Li et al., 2010). La cascata ERK5/KLF4 può agire come perno in vari percorsi che proteggono i neuroni dalla morte indotta da stress ossidativo(Su et al., 2014).
Lo stress ossidativo è stato considerato strettamente correlato a molte malattie degenerative. KLF4 svolge un ruolo significativo nel mantenimento della stabilità genomica nello stress ossidativo. KLF4 e ERK5 agiscono insieme per proteggere i neuroni dall’apoptosi indotta dallo stress ossidativo. Pertanto, KLF4 può agire come bersaglio terapeutico per agire contro lo stress ossidativo quando attivato. È stato riportato che i farmaci statinici possono attivare ERK5, portando all’espressione di KLF4 e dei suoi geni dipendenti (Ohnesorge et al., 2010), ma il meccanismo rimane poco chiaro, e i geni target a monte e a valle di KLF4 sono meno studiati nello stress ossidativo, c’è bisogno di ulteriori studi.
Ruolo del KLF4 nella rigenerazione degli assoni
La perdita precoce degli assoni è una caratteristica comune delle malattie neurodegenerative. La perdita sinaptica e la compromissione del trasporto nell’AD possono causare disturbi cognitivi (Holtzman et al., 2011; Coleman, 2013). Il grado di danno dichiarativo alla memoria è correlato alla densità sinaptica nell’ippocampo e nella corteccia. Gli oligomeri solubili di Aß riducono l’assorbimento del glutammato e promuovono la disfunzione sinaptica, interrompendo la plasticità sinaptica (Li et al., 2009). Pertanto, è particolarmente importante studiare come riparare gli assoni nel SNC. Nelle cellule gangliari retiniche, gli assoni hanno una forte capacità di crescere e rigenerarsi durante lo sviluppo precoce, ma nel SNC dei mammiferi adulti, gli assoni perdono la loro capacità di rigenerazione e i neuroni possono laurearsi per morire o atrofizzarsi (Goldberg e Barres, 2000; Goldberg et al., 2002).
KLF4 svolge un ruolo importante nell’inibizione della crescita degli assoni. Nei RGCS embrionali, la sovraespressione di KLF4 può ridurre la percentuale di allungamento dei neuriti, la lunghezza degli assoni e dei dendriti e la ramificazione dei neuriti. Inoltre, è stato riscontrato che la sovraespressione di KLF4 può ridurre i tassi di crescita degli assoni postnatali a lungo termine, ma non è riuscita a ridurre i tassi di crescita degli assoni a breve termine (Moore et al., 2009; Steketee et al., 2014). Studi successivi hanno scoperto che i fasci di assoni dei topi KLF4–cKO erano più spessi dei topi di controllo (Fang et al., 2016). Inoltre, la rimozione dell’espressione KLF4 durante lo sviluppo può aumentare il potenziale riproduttivo degli RGCS adulti. Inoltre, KLF4 privo del dominio di legame del DNA c-terminale non ha avuto alcun effetto sulla crescita degli assoni. Non c’è stato alcun impatto sulla sopravvivenza delle cellule dopo che le cellule gangliari retiniche sono state ferite se il KLF4 è stato eliminato (Moore et al., 2009).
KLF4 può anche influenzare la rigenerazione assonale. Uno studio recente ha riferito che la diminuzione dell’espressione di KLF4 nelle cellule gangliari retiniche adulte ha promosso la rigenerazione degli assoni attraverso la via JAK-STAT3 (Qin et al., 2013). KLF4 ha aumentato la fosforilazione di STAT3 e ha regolato la crescita degli assoni tramite la segnalazione JAK-STAT (Qin e Zhang, 2012). Nell’ambito del trattamento delle citochine, i membri della famiglia di STAT delle proteine sono fosforilati ai siti carbossi-terminali della tirosina e della serina all’interno della cellula per formare un dimero stabile. Questa modifica migliora la trascrizione dei geni associati alle cellule (Yuan et al., 2005). L’interazione tra KLF4 e STAT3 sulla fosforilazione indotta da citochine di tirosin705 inibisce l’espressione di STAT3 inibendo il legame di STAT3 al DNA (Qin et al., 2013). KLF4 knockdown ovviamente migliora la rigenerazione di assone in cellule gangliari retinici dopo la lesione del nervo ottico, e impedisce il nervo da lesioni dopo lesioni cerebrali lievi. Le azioni sono mediate da una diminuzione di p-p53 e un aumento dei livelli di pSTAT3. KLF4 regola positivamente l’apoptosi neuronale tramite le vie p53 e JAK-STAT3 e KLF4 regola negativamente la riparazione assonale tramite la via JAK-STAT3 (Cui et al., 2017).
Pertanto, abbiamo ipotizzato che in AD, la rigenerazione assonale possa essere ottenuta alterando l’espressione di KLF4 o alterando le vie di segnalazione correlate intracellulari e controllando la progressione dell’AD riducendo gli assoni mancanti o riducendo la disfunzione assonale. Tuttavia, come utilizzare il fattore di trascrizione KLF4 in potenziali terapie ha ancora bisogno di ulteriori esplorazioni.
Ruolo di KLF4 nell’accumulo di ferro
Il ferro è ampiamente presente nei sistemi biologici, le metalloproteinasi correlate al ferro svolgono un ruolo chiave nel trasporto di ossigeno, nel trasferimento di elettroni e nella catalizzazione delle reazioni biochimiche (Aisen et al., 2001). Tuttavia, qualsiasi eccesso di ferro oltre il normale intervallo fisiologico può danneggiare la salute umana (Adlard e Bush, 2006). Gli studi hanno scoperto che il contenuto di ferro nell’ippocampo è negativamente correlato con le prestazioni dei test di memoria (Ding et al., 2009). L’aumento del carico di ferro nel cervello accelera la formazione di placche Aß e grovigli tau iperfosforilati, migliorando allo stesso tempo lo stress ossidativo (Peters et al., 2015). Il ferro, che ha un alto grado di permeabilità, promuove la crescita nervosa e le connessioni da cellula a cellula durante lo sviluppo del cervello (Dallman e Spirito, 1977).
Uno studio recente ha dimostrato che lo stress fisiologico ha causato l’attivazione della via di segnalazione KLF4-HCP1 e un aumento dell’assorbimento dell’eme (Li H. et al., 2017). L’eme rappresenta il 95% del ferro funzionale nel corpo umano. È uno dei componenti principali dell’eme ossigenasi (Hooda et al., 2014; Kurucz et al., 2018). Aumentare l’attività dell’ossigenasi – 1 può ritardare l’ossidazione del cervello che invecchia (Verdile et al., 2015; Serini e Calviello, 2016; Kurucz et al., 2018). Questo ha un effetto di sollievo su AD. Lo sforzo fisiologico induce l’aumento livellato glucocorticoide, glucocorticoide aumenta l’espressione della proteina 1 (HCP1) del trasportatore di heme via KLF4 e poi HCP1 promuove l’assorbimento di heme (Li H. et al., 2017). Glucocorticoidi e KLF4 regolano insieme i geni anti-infiammatori e le cellule con basso contenuto di glucocorticoidi non possono indurre completamente l’espressione di KLF4 (Sevilla et al., 2015). L’aumento indotto da KLF4 nell’assunzione di eme porta all’accumulo di ferro nel cervello. Il ferro promuove il rilascio di ROS (Tronel et al., 2013). L’elemento di ferro aumenta lo stress ossidativo cerebrale nei ratti sotto stress psicologico (Yu et al., 2011). Pertanto, HCP1 può essere regolato da KLF4 e glucocorticoide insieme. L’aumento dell’HCP1 aumenta l’assorbimento dell’eme, che porta direttamente all’accumulo di ferro nel cervello, esacerba l’ossidazione, aumenta l’apoptosi o la disfunzione e peggiora il danno cerebrale.
È generalmente accettato che la memoria e la disabilità di apprendimento siano i principali sintomi dell’AD. Un gran numero di dati clinici hanno dimostrato che Aß carico placca e accumulo di ferro risposta allo sviluppo di apprendimento e disfunzione cognitiva in AD (van Bergen et al., 2018). I dati pubblicati di recente hanno suggerito che il ferro ad alte dosi aumenta la deposizione di Aß e attenua l’apprendimento e la memoria nei topi (Guo et al., 2013). Studi clinici hanno dimostrato che la microglia contenente ferro si trova nell’ippocampo di pazienti con AD sotto risonanza magnetica (Zeineh et al., 2015). La microglia acquisisce ferro da fonti trasferenti o non trasferenti, extracellulari e intracellulari (McCarthy et al., 2018). L’espressione selettiva e sostenuta di KLF4 può essere indotta nel nucleo e nel citoplasma degli astrociti reattivi dell’ippocampo ischemico (Park et al., 2014). Gli studi hanno dimostrato che KLF4 agisce come un repressore trascrizionale. Si abbassa-regola l’espressione di ELK-3, e quindi ELK-3 inibisce l’espressione di HO-1 (Tsoyi et al., 2015). Eme ossigenasi – 1 (HO-1) è una proteina di stress che degrada eme in bilirubina, ferro libero e monossido di carbonio. L’up-regulation di HO-1 negli astrociti può portare a depositi anormali di ferro e disfunzione mitocondriale nel cervello, portando a una diminuzione della capacità cognitiva (Schipper, 1999, 2004). Pertanto, KLF4 può essere coinvolto nel processo di accumulo di ferro negli astrociti, esacerbando l’ossidazione nell’AD e aggravando il danno cerebrale.
Conclusione
KLF4 è comunemente noto per svolgere un ruolo fondamentale nella regolazione della proliferazione cellulare, dell’apoptosi e della differenziazione. Studi precedenti si sono concentrati sulla regolazione del KLF4 in diversi importanti processi neurofisiologici, tra cui neuroinfiammazione, neuroprotezione e rigenerazione sinaptica. Recentemente, KLF4 è stato trovato per svolgere un ruolo importante nella patogenesi di AD. In questo articolo, esaminiamo il ruolo di KLF4 nella neuroprotezione e nella neurogenesi nell’AD.
KLF4 non è solo un regolatore di regolazione della proliferazione e differenziazione cellulare, ma anche un potenziale bersaglio per la regolazione delle risposte immunitarie. KLF4 può regolare i fattori infiammatori negativi e promuovere la risposta infiammatoria e avere un grande effetto sull’espressione della microglia nucleare dell’astrocita. Inoltre, KLF4 e ERK5 possono agire insieme per esercitare azioni neuroprotettive. Inoltre, la rigenerazione degli assoni può essere ottenuta alterando il contenuto di specifici fattori di trascrizione, inibitori intracellulari o alterando le vie di segnalazione intracellulari. Eliminare KLF4 può migliorare la rigenerazione degli assoni e accelerare il tasso di crescita degli assoni. La riduzione dell’espressione di KLF4 promuove la rigenerazione degli assoni attraverso la via JAK-STAT3 e KLF4 promuove la via JAK-STAT3 per un’ulteriore rigenerazione degli assoni. Pertanto, KLF4 potrebbe essere coinvolto nel processo di anti-infiammatori, anti-apoptosi, rigenerazione degli assoni e accumulo di ferro nel SNC, che svolge un ruolo fondamentale nella generazione di AD. Questi risultati suggeriscono che KLF4 rappresenta un potenziale bersaglio terapeutico per l’AD. Tuttavia, i meccanismi cellulari e molecolari profondi degli effetti di KLF4 sulla AD rimangono poco chiari e sono necessarie ulteriori indagini.
Contributi dell’autore
ZQC, XHZ e YJ hanno scritto il manoscritto. SHG e JYL hanno modificato il quadro del manoscritto. BJL e RJC hanno fornito le revisioni critiche. Tutti gli autori hanno approvato la versione finale del manoscritto per la presentazione.
Finanziamento
Questo lavoro è stato sostenuto da sovvenzioni della Natural Science Foundation of China (NSFC) (81871070, 31571126, 31471120) e Jilin Science and Technology Agency funding (Grant Nos. 20180519003JH, 20180414051GH, 20170414034GH e 20180414050GH).
Dichiarazione sul conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di relazioni commerciali o finanziarie che potrebbero essere interpretate come un potenziale conflitto di interessi.
Cacabelos, R. (2018). Ci sono stati miglioramenti nella scoperta di farmaci per la malattia di Alzheimer negli ultimi 5 anni. Scad. Opin. Droga Discov. 13, 523–538. doi: 10.1080/17460441.2018.1457645
PubMed Abstract / CrossRef Testo completo / Google Scholar
Liu, Y., Shi, J. e Chen, X. (2015). Identificazione di nuovi bersagli per la rinite allergica stagionale durante e al di fuori della stagione dei pollini mediante analisi microarray. Acta Otorinolaringoiatria. 135, 1330–1336. doi: 10.3109 / 00016489.2015.1067906
PubMed Abstract / CrossRef Testo completo / Google Scholar
Serini, S., e Calviello, G. (2016). Riduzione dello stress ossidativo/nitrosativo nel cervello e il suo coinvolgimento nell’effetto neuroprotettivo di n-3 PUFA nella malattia di Alzheimer. Curr. Alzheimer Res. 13, 123-134. doi: 10.2174/1567205012666150921101147
PubMed Abstract / CrossRef Full Text / Google Scholar