La tecnologia per il sequenziamento del DNA è stata sviluppata nel lontano 1977 grazie a Frederick Sanger. Ci è voluto un po ‘ di più prima che fosse possibile sequenziare un genoma completo. Questo perché avevamo bisogno di un modello matematico appropriato e di una massiccia potenza computazionale per assemblare milioni o miliardi di piccole letture su un genoma completo più grande. La potenza computazionale e il software di oggi sono la principale differenza tra ciò che richiedeva anni di lavoro nei primi anni 2000 e ciò che richiede solo poche ore oggi. L’algoritmo che hai scelto per farlo è il “santo graal” della tecnologia di assemblaggio. Questi algoritmi incorporano una delle variabili più famose conosciute nei modelli matematici, la k-mer.
L’origine del k-mer e del modello matematico che lo circonda proviene da un matematico svizzero del 1735 Leonhard Euler, che è noto come il padre della funzione matematica. Un matematico olandese Nicolaas de Bruijn adattò le idee di Eulero per trovare una sequenza ciclica di lettere prese da un dato alfabeto per cui ogni possibile parola di una certa lunghezza appare come una stringa di caratteri consecutivi nella sequenza ciclica esattamente una volta.
l’algoritmo di de Bruijn è stato adattato dai biologi molecolari, che molti anni dopo hanno affrontato un problema equivalente: come assemblare sequenze di DNA. Pertanto, gli scienziati di tutto il mondo ora usano il grafico De Bruijn e la variabile k.
Applicazione di k-mers all’assemblaggio di sequenze di DNA
In poche parole, l’assemblaggio del genoma de novo comporta il collegamento di piccole letture consecutive del DNA e termina con sequenze più grandi. Per generare un grafico di de Bruijn (vedi la figura sotto), i nucleotidi sul bordo di ogni lettura devono sovrapporsi al bordo di una seconda (e così via). L’obiettivo finale è quello di creare un vertice consecutivo, che (potenzialmente) si tradurrà in grandi frammenti di DNA.
Devi frammentare le tue letture in k-mer, che sono un numero specifico di nucleotidi che si sovrappongono. Il k-mer consente di generare una sequenza unica da molti piccoli. Ogni sequenza k-mer univoco viene identificato e copie extra vengono eliminati. Questo aspetto di k-mers consente di superare uno degli svantaggi del sequenziamento di prossima generazione: ottenere letture che rappresentano regioni genomiche con frequenze diverse (cioè, ottenere molte piccole letture da una regione). L’uso di k-mers elimina le sequenze ripetute più di una volta a causa della copertura disuguale della sequenza. Tuttavia, tieni presente che una bassa dimensione k-mer aumenterà le possibilità di sovrapposizione dei nucleotidi, pur avendo un valore maggiore li diminuirà.
La tecnologia di assemblaggio de novo di oggi è più efficiente quando si utilizzano librerie di letture di grandi dimensioni (ad esempio, 1.000–10.000 bps) combinate con quelle più piccole (100-200 bps). I programmi software possono utilizzare il valore k e k-mers per assemblare brevi letture. Questi possono quindi essere incorporati e verificati da quelli più grandi per finire in contig più accurati.
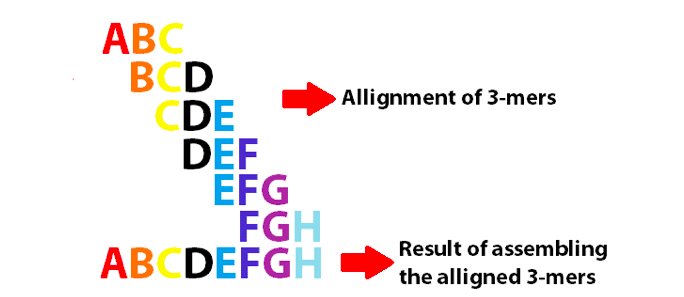
Esempio di un grafico de Bruijn che utilizza 3-mers per assemblare le 8 prime lettere dell’alfabeto inglese. Si noti che questi 3-mers si sovrappongono come k-1.
Più ne sai e più puoi ottenere nell’assemblaggio del DNA
Ci sono suggerimenti specifici che devi considerare prima di applicare i grafici De Bruijn nel tuo metodo di assemblaggio e scegliere la dimensione k-mer più appropriata. Sfruttando questi, è possibile generare risultati migliori.
- Prima di tutto, e forse la cosa più importante, è usare molti diversi k-mer nel tuo assembly. Si dovrebbe quindi valutare i risultati e scegliere il migliore (s). Non dimenticare mai che non c’è quasi mai uno e solo un assemblaggio corretto.
- È necessario gestire attentamente le letture degli errori, prima di utilizzare un k-mer. Se non si rimuovono con attenzione gli errori, i risultati possono creare un rigonfiamento indesiderato, complicando l’assemblaggio. Aumentare la soglia per il tasso di errore utilizzato durante il ritaglio della sequenza. Potresti perdere alcune sequenze, ma quelle che rimangono saranno le più belle.
- È necessario gestire con attenzione le ripetizioni del DNA. Ad esempio, Illumina sequencing genera una grande quantità di dati. Innanzitutto, prova a assemblare una piccola frazione delle letture e poi usale tutte per individuare le differenze. Le letture brevi ripetibili potrebbero interferire negativamente con il processo di assemblaggio.
- Conosci i tuoi dati. Se non si conosce la dimensione del vostro genoma previsto, la quantità di copertura sequenziamento, e il numero di letture, allora siete più inclini a scegliere il miglior valore k per assemblare il genoma. Puoi visitare k-mer advisors, come velvet advisor della Monash university per avere qualche consiglio su quale valore sembra più adatto.
L’uso di k-mer di varie lunghezze e l’allineamento dei contig aiuta anche i ricercatori a individuare i tassi di mutazione, espandendo il suo uso. Naturalmente, manipolare i grafici De Bruijn verso il beneficio dell’assemblaggio non è una panacea. Ci sono molte cose da considerare che una funzione semplicistica per assemblare il genoma di un organismo vivo. Questa è solo un’introduzione della storia e di come i biologi possono usarlo in modo più efficiente.
- Compeau PE, Pevzner PA, Tesler G. (2011). Come applicare i grafici de Bruijn all’assemblaggio del genoma.Biotecnologie naturali. 29(11):987–91.
- Aggarwala V, Voight BF. (2016). Un modello di contesto sequenza espansa spiega ampiamente variabilità nei livelli di polimorfismo in tutto il genoma umano. Natura Genetica. 48(4): 349–55.

Questo ti ha aiutato? Quindi si prega di condividere con la rete.