はじめに
Kruppel様因子4(KLF4)は、様々なヒト組織で発現される亜鉛指転写因子のファミリーのメンバーである。 これは、多能性幹細胞(iPSC)への誘導の4つの因子の1つとしてよく知られている(Ghaleb and Yang、2017)。 KLF4は、神経炎症、酸化ストレス、増殖、分化、およびアポトーシスのような複数の重要な生物学的プロセスを調節することができる(Kaushik e t a l. ら、2 0 1 0;Mamonkin e t a l. ら、2 0 1 3;Zhang e t a l. ら、2 0 1 5;Miao e t a l. 2017年;Xu,et al., 2017). 以前の研究の量は、癌の発生および進行におけるKLF4の役割に焦点を当てた(Karam e t a l. ら、2 0 1 7;Yadav e t a l., 2018). KLF4は二重機能転写因子であり、癌の種類または癌の病期に応じて癌遺伝子または腫瘍抑制遺伝子としての役割を発揮することができる(Evans and Liu,2 0 0 8)。 それは、細胞増殖、分化およびアポトーシスに関与する遺伝子の転写を活性化または阻害することができる(Ding et al., 2015). KLF4は、他の初期化因子と協力して体細胞をiPSCに変換し、幹細胞の分化を阻害することができる(Takahashi and Yamanaka,2 0 0 6;van Schaijik e t a l., 2018). これは、血管疾患、免疫疾患、食欲不振および他の疾患の治療的見通しを提供する(Imbernon et al. ら、2 0 1 4;Liu Y. ら、2 0 1 5;Murgai e t a l., 2017). さらに、KLF4は、中枢神経系(CNS)において広く調節的役割を果たすことができる。 いくつかの研究は、KLF4が、アルツハイマー病(A D)、てんかん、パーキンソン病、水頭症および統合失調症を含む、複数の神経障害に関連していることを示している(Qin e t a l. ら、2 0 1 1;Xie e t a l. ら、2 0 1 3;Han e t a l. ら,2 0 1 5;Nishiguchi e t a l. ら、2 0 1 5;Li L., 2017).
ADは最も一般的な慢性神経変性疾患の一つであり、認知および記憶障害、様々な精神症状および行動異常をもたらし、進行性認知症が最も一般的な臨床的特徴である(Jiang et al., 2018). ADの現在確認されている病原因子には、異常なアミロイド−β(A Β)沈着によって誘導される老人性プラークの形成、およびタウ蓄積によって誘導される神経原線維のもつれまたはジストロフィー神経炎が含まれる(Querfurth and Laferla,2 0 1 0;Shinohara e t a l., 2014). さらに、ADは遺伝的要因によっても影響を受ける可能性があります。 しかし、誘発された病因はまだ不明である。 AD治療のための最も一般的な薬物には、神経伝達物質エンハンサー、抗アミロイド剤、神経保護ペプチド、および他の薬物が含まれる(Cacabelos、2018)。 特に、いくつかの研究では、KLF4がADの病因において重要な役割を果たしたことが示されている。 このレビューでは、神経炎症、神経アポトーシス、軸索再生、およびKLF4と神経変性疾患の細胞および分子メカニズムへの洞察を提供するかもしれないADの病因との間の関連付けを説明するために鉄蓄積におけるKLF4の調節役割に焦点を当てています。
KLF4の生物学的特性
KLF4は、NIH3T3ライブラリから単離され、細胞核に位置する亜鉛フィンガー含有核タンパク質である。 最初に同定され、Shieldsらによって特徴づけられた。 (1996). ヒトKLF4の分子量は55kdであり、染色体9q31に位置する。 KLF4は6.3kbの遺伝子セグメントをカバーし、五つのエクソンを持っています。 そのcDNAコード領域は、4 7 0個のアミノ酸残基からなるポリペプチドをコードする(Yet e t a l. ることを示した。 KLF4のカルボキシ末端は、81高度に保存されたアミノ酸によって形成されている三つのCys2His2(C2H2)型亜鉛指構造を含むDNA結合構造領域を有する。 それは、CACCC要素およびGCに富む標的遺伝子DNA配列との高親和性によって転写を調節する(ShieldsおよびYang,1 9 9 8;Pearson e t a l., 2008). KLF4のDNA結合部位の大部分は、相互作用するタンパク質のためのN末端転写活性化ドメイン、DNA結合のためのC末端亜鉛指構造および転写阻害ゾーン(Bieker,2 0 0 1) KLF4は、多くの内因性遺伝子の発現の調節に関与している(Shields and Yang,1 9 9 8)。 KLF4のアミノ末端に非常に可変転写調節ドメインがあります。 91と117アミノの間に位置するアミノ酸残基は、転写抑制ドメインも存在するが、プロリンとセリンが豊富な転写活性化ドメインを構成する。 したがって、KLF4は、遺伝子転写を活性化および阻害する2つの有害作用を有する(Yet e t a l. 1998年;Wei et al., 2006).
胚発生中、KLF4は胚発生の後期段階でより高く発現していた。 成熟した組織および器官では、KLF4は主に消化管、口腔、皮膚表皮、血管内皮および腎臓で発現され、脳では発現されない(Segre e t a l. ら、1 9 9 9;Ghaleb e t a l. ら、2 0 1 1;Liu e t a l. ら、2 0 1 3;Chen e t a l. 2015年、He et al. ら、2 0 1 5;Bin e t a l., 2016). これは、細胞増殖および分化の調節に重要な役割を果たすと考えられている。 その上、KLF4はまた細胞周期を調整できます。 KLF4は、P5 3依存的にP2 1を活性化することができる(Zhang e t a l., 2000). さらに、KLF4(–/–)細胞はKLF4(+/+)細胞よりも早く老化期に入ったことが判明し、KLF4(–/–)細胞における抗酸化遺伝子発現が少なく、活性酸素種(ROS)レベルが高いことによ ROSは、p5 3およびp2 1発現を増加させ、続いてDNA損傷を促進することができる(Liu C.et a l., 2015). これは、PRMT5は、タンパク質レベルでKLF4発現を上昇させることができることが判明しました。 PRMT5は、KLF4ユビキチン化を阻害することにより、p21の転写を増加させ、baxの発現を減少させることが報告された(Hu et al., 2015). さらに、多数の研究は、KLF4がニューロンのアポトーシスの調節に関与していることを実証している(Kong e t a l. ら、2 0 1 6;Cui e t a l. 2017年、Song et al., 2018). 我々が知られているKLF4の生理学的調節役割はまだほとんどなく、さらなる調査が必要である。
ADにおけるKLF4の役割
ADは主に記憶障害および認知障害および執行機能不全によって特徴付けられることは十分に確立されている(Goedert and Spillantini、2006)。 多くの研究は、神経細胞のアポトーシスおよびシナプス機能不全が、認知機能の低下の病理学的基礎であることを実証している(Caccamo et al. ら、2 0 1 7;Guo e t a l. ら、2 0 1 7;Yoon e t a l., 2018). A Β沈着、酸化ストレスおよび鉄蓄積の蓄積された損傷は、AD患者の神経機能障害およびアポトーシスにつながる可能性がある。 いくつかの研究では、KLF4の調節役割がCNSにおいて重要であると思われることが示されている。 KLF4は、神経アポトーシス、シナプス再生、酸化ストレスと神経炎症を調節することが報告されたことを考慮すると、KLF4とADの病因との関係は、AD治療のた
神経炎症におけるKLF4の役割
臨床研究の量は、A Βが凝集し、AD患者の脳組織の細胞外沈着物の主成分であり、周囲のシナプスやニューロンを損 A Βの異常な分泌または過剰な産生はADの病理学的変化をもたらすので、A Β沈着はADの中核的なリンクである(Rajmohan and Reddy、2017)。 さらに、研究では、過剰なA Β沈着がグリア細胞を刺激してROSおよび他の影響因子を分泌し、酸化ストレスを引き起こすことが示されている。 酸化ストレスはA Βの産生を刺激することが知られていた。 従って、A Βおよび酸化ストレスは、互いに相互作用し、ADの進行に影響を及ぼすことができる(Cheignon e t a l., 2018).
KLF4は潜在的なモジュレーターとして報告されており、マクロファージや内皮細胞を媒介することによって炎症に大きな影響を与える(図1)(Yoshida et al. る。 ら、2 0 1 5;Yang e t a l., 2018). 中枢神経系では、過剰および慢性の炎症反応は、ニューロンおよび神経細胞の損傷を引き起こす可能性がある。 これは、最近KLF4発現が正のΒ42誘導神経炎症と相関することが実証されました。 ミクログリアBV2細胞では、オリゴマー A Β42は、活性化されたP53によって媒介されるKLF4発現を増加させることができる(Li L.et al., 2017). A Β蓄積のような炎症性条件下では、炎症促進性サイトカインの放出は、ADの生成において刺激され得る(Griffin and Barger,2 0 1 0)。 可溶性A Β4 2オリゴマーの神経毒性効力および炎症促進効力は、不溶性繊維沈着物よりも比較的高い(Selkoe,1 9 9 1;Weinberg e t a l.,1 9 9 1;Weinberg e t a l.,1 9 9 2;Weinberg e t a l., 2018). KLF4の沈黙は、Β4 2媒介性神経炎症を回復することができ、KLF4の過剰発現は、Β4 2媒介性神経炎症を悪化させることができる(Li L.et al., 2017). A β蓄積は、アストロサイトおよびミクログリアの活性化を誘導する(Rodriguez e t a l., 2016). 活性化されたアストロサイトは、IL−1、IL−6、およびTNF−αなどの炎症促進因子を放出することによって神経炎症を増強することができる(Rubio−PerezおよびMorillas−Ruiz、2 0 1 2;Domene e t a l., 2016). 炎症性応答の悪循環は機能障害および神経のapoptosisの結局導きます。
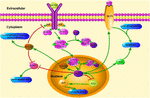
図1. KLF4関連シグナル伝達経路のSchematicillustration。 この図は、神経保護と軸索再生におけるKLF4の役割を強調しています。 図中の矢印は活性化または促進を示し、直線は関連する阻害を示す。 KLF4、Kruppel様因子4;stat3、転写シグナル変換器および活性化因子3;JAK、Janusキナーゼ;socs3、サイトカインシグナル伝達の抑制因子3;HCP1、ヘムキャリアプロテイン1;ERK5、
KLF4は、プロ炎症性シグナルの調節に重要な役割を果たしています。 グリア細胞では、ゲムフィブロジル誘導KLF4活性化は、PI3-キナーゼ-AKT経路を介してサイトカインシグナル3(SOCS3)のサプレッサーを増加させる(Ghosh and Pahan、2012)。 KLF4のSirna媒介ノックダウンは、マウスのアストログリアおよびミクログリアにおけるSOCのレベルを減弱させることができ、これは、続いて炎症性遺伝子の発現に影響を及ぼす可能性がある(Kaushik e t a l. ることを示した。 さらに、SOCS欠失は、損傷したニューロンの生存を促進し、軸索再生を促進することができる(Smith e t a l. ら、2 0 0 9;Sun e t a l., 2011). そして、KLF4は、IL-1βまたは他の炎症促進マーカーの産生を積極的に調節する。 それは積極的にシクロオキシゲナーゼ-2(Cox-2)を調節し、誘導性一酸化窒素シンターゼ(iNOS)を負に調節する(Kaushik et al., 2013). さらに、KLF4は、単球分化のための重要な調節因子であり、免疫調節のための潜在的な標的である(Alder e t a l., 2008). 従って、KLF4はこれらの否定的な調整装置の調整によってneuroinflammationを促進するかもしれません。
パーキンソン病モデルでは、KLF4はMPP+誘発性酸化ストレスおよび神経毒性を促進し、神経アポトーシスを増加させ、細胞増殖を遅らせることができることに言及する価値がある(Chen et al., 2013). 酸化ストレスは、過酸化と抗酸化の間の不均衡である。 フリーラジカルは、異なる高分子の変化を引き起こし、細胞の損傷、細胞の老化および組織の損傷を引き起こす可能性がある(Parajuli et al. ら、2 0 1 3;Nie e t a l., 2015). 酸化ストレスは、早期の炎症およびA Β産生を悪化させ、次いでADを悪化させることができる(Cai e t a l., 2011). したがって、KLF4は、ADの酸化ストレスに関与している可能性があります。
これらの知見は、KLF4がミクログリアを活性化し、結果的に炎症性サイトカインの放出によって神経炎症を媒介する重要な役割を示唆している。 それは神経炎症を強化する可能性があります。 これまでのところ、ADの病因に関する多くの研究は、神経炎症に焦点を当ててきました。 免疫調節のための潜在的な標的として、KLF4は、関連する負の調節因子に影響を与えることを介してミクログリアの炎症反応を促進することができ、これはADの発症に大きな影響を及ぼす。
アポトーシスにおけるKLF4の役割
神経変性変化には、大脳皮質、海馬および他の皮質下領域などの代表的な脳領域におけるニューロンおよびシナプス 神経細胞の喪失によって誘導される中枢神経系の機能障害は永続的である(Citron、2010)。 持続的な酸化ストレスは、神経細胞のアポトーシスにつながる可能性がある(Wu et al., 2010). 多数の研究により、ADが酸化ストレスと密接に関連していることが確認されている(Lee e t a l.,2012;Yui et al., 2015). 慢性酸化ストレスは、アストロサイトにおけるホスホリパーゼA2グループ3(Pla2G3)の発現を増強し、A Βのバランスを破壊し、結果的にADの発症につな, 2015).
多くの研究が、KLF4が酸化ストレスの発生を抑制する上で重要な役割を果たすことを実証している(Shi et al. ら、2 0 1 4;Liu C., 2015). KLF4がH2O2によって誘導される細胞のアポトーシスを促進できることが見出され、この作用はbax発現の増加およびbcl-2発現の減少によって引き起こ, 2010). ケルセチンは、ヒト神経芽細胞腫SH-SY5Y細胞におけるKLF4発現を減少させ、bcl-2/bax比の発現を増加させることができる。 さらに、ケルセチンは、SH-5YSY細胞のアポトーシス速度を緩和し、カスパーゼ-3酵素活性を低下させることができる(Xi et al., 2012). 最近の研究では、酸化ストレスに対するマイトジェン活性化タンパク質(MAP)キナーゼ5(ERK5)の神経保護効果を調べた。 ERK5の活性化は、H2O2誘導性の海馬ニューロンの死を部分的に減少させ、NGFおよびPC誘導性の神経保護を増加させることができる(Su e t a l., 2014). Nils et al. ヒト内皮細胞におけるERK5活性化転写および機能的応答を研究するためにMEK5の変異体(MEK5D)を使用し、KLF4が新規な下流ERK5標的であることを, 2010). これは、KLF4の過剰発現は、TNFを介した炎症反応を抑制し、白血球の接着と基底細胞アポトーシスを減少させることができることが見出された。 これらの結果は、KLF4が抗炎症および抗アポトーシス特性を有することを確認する(Ohnesorge e t a l., 2010). その後の実験は、内皮細胞における脳海綿体奇形1(CCM1)の消失が、MEKK3−MEK5シグナル経路を介してERK5を活性化し、KLF4発現を増加させることを実証した(Cuttano e t a l., 2016). ERK5は、前処理(PC)および神経成長因子(NGF)において仲介的役割を果たし、KLF4の発現を上方調節した(Su e t a l., 2014). さらに、KLF4のRnai媒介ノックダウンは、NGFまたはPC誘導性神経保護を減少させることもできる。 KLF4の過剰発現は、H2O2ストレス細胞においてより高いbcl−2/bax比をもたらす(Su e t a l., 2014). 過剰発現KLF4は、その対応するプロモーターと結合することにより、bcl−2およびbaxの変化を促進する(Li e t a l., 2010). ERK5/KLF4カスケードは、酸化的ストレス誘発死からニューロンを保護する様々な経路のピボットとして作用し得る(Su e t a l., 2014).
酸化ストレスは多くの変性疾患と密接に関連していると考えられている。 KLF4は、酸化ストレスにおけるゲノム安定性を維持する上で重要な役割を果たしている。 KLF4およびERK5は酸化圧力誘発のapoptosisからニューロンを保護するために一緒に機能します。 従って、KLF4は活動化させたとき酸化圧力に対して機能するために治療上のターゲットとして機能するかもしれません。 スタチン薬がERK5を活性化し、KLF4およびその依存遺伝子の発現をもたらすことが報告されている(Ohnesorge e t a l.,2010),しかし、メカニズムは不明のまま,そしてKLF4関連上流および下流の標的遺伝子は、酸化ストレスではあまり研究されていません,さらなる研究の必要
軸索再生におけるKLF4の役割
初期の軸索喪失は神経変性疾患の共通の特徴である。 ADにおけるシナプス損失および輸送障害は、認知障害を引き起こす可能性がある(Holtzman e t a l. 2011年、コールマン、2013年)。 宣言的記憶損傷の程度は、海馬および皮質におけるシナプス密度に関連している。 可溶性A Βオリゴマーは、グルタミン酸の取り込みを減少させ、シナプス機能障害を促進し、シナプス可塑性を破壊する(Li e t a l., 2009). したがって、中枢神経系の軸索を修復する方法を研究することは特に重要である。 網膜神経節細胞では、軸索は、初期の発達の間に成長し、再生する強力な能力を有するが、成体哺乳動物のCNSでは、軸索は、その再生能力を失い、ニューロンは、死, 2002).
KLF4は軸索の成長を阻害する上で重要な役割を果たしています。 胚RGCsでは、KLF4の過剰発現は、神経突起伸長の割合、軸索および樹状突起の長さ、および神経突起分岐を減少させることができる。 さらに、KLF4の過剰発現は、出生後の長期軸索成長率を低下させることができるが、短期軸索成長率を低下させることができないことが見出された(Moore e t a l. ら,2 0 0 9;Steketee e t a l., 2014). 後の研究は、KLF4−cKOマウスの軸索束が対照マウスよりも厚いことを見出した(Fang e t a l., 2016). さらに、発達中のKLF4発現の除去は、成体Rgcの生殖可能性を増加させることができる。 さらに、C末端DNA結合ドメインを欠いているKLF4は、軸索の成長に影響を与えなかった。 KLF4がノックアウトされた場合、網膜神経節細胞が損傷した後の細胞の生存に影響はなかった(Moore e t a l., 2009).
KLF4は軸索再生にも影響を与える可能性があります。 最近の研究では、成体網膜神経節細胞におけるKLF4発現の減少が、JAK−STAT3経路を介した軸索再生を促進することが報告されている(Qin e t a l., 2013). KLF4はSTAT3のリン酸化を増加させ、JAK-STATシグナル伝達を介して軸索の成長を調節した(Qin and Zhang、2012)。 サイトカインの処理の下で、タンパク質のSTATファミリーのメンバーは、安定した二量体を形成するために、細胞内のカルボキシ末端チロシンおよびセリン この修飾は、細胞関連遺伝子の転写を増強する(Yuan et al., 2005). チロシン705のサイトカイン誘発性リン酸化上のKLF4とSTAT3との間の相互作用は、DNAへのSTAT3の結合を阻害することにより、STAT3の発現を阻害する(Qin et al., 2013). KLF4ノックダウンは明らかに視神経の傷害の後で網膜の神経節の細胞の軸索の再生を改善し、穏やかな脳損傷の後で傷害から神経を防ぎます。 この作用は、p-p53の減少およびpstat3レベルの増加によって媒介される。 KLF4は、p5 3およびJAK−STAT3経路を介して神経細胞のアポトーシスを積極的に調節し、KLF4は、JAK−STAT3経路を介して軸索修復を否定的に調節する(Cui e t a l., 2017).
したがって、我々は、ADでは、KLF4の発現を変化させるか、細胞内関連のシグナル伝達経路を変化させ、不足している軸索を減少させるか、軸索機能障害を減 しかし、KLF4転写因子を潜在的な治療法にどのように使用するかは、さらなる研究が必要である。
鉄蓄積におけるKLF4の役割
鉄は生物学的系に広く見られ、鉄関連メタロプロテイナーゼは酸素の輸送、電子の移動、生化学反応の触媒に重要な役割を果たす(Aisen et al., 2001). しかし、通常の生理学的範囲を超える鉄の過剰は、人間の健康を損なう可能性がある(Adlard and Bush、2006)。 研究は、海馬中の鉄含有量が記憶試験の性能と負の相関があることを見出した(Ding et al., 2009). 脳内の鉄負荷の増加は、A Βプラークおよび過リン酸化タウのもつれの形成を加速し、同時に酸化ストレスも増強する(Peters e t a l., 2015). 高度の透過性を有する鉄は、脳の発達中に神経の成長および細胞間の接続を促進する(DallmanおよびSpirito、1977)。
最近の研究では、生理学的ストレスがKLF4-HCP1シグナル伝達経路の活性化を引き起こし、ヘム取り込みを増加させることが示された(Li H.et al., 2017). ヘムは人体の機能性鉄の95%を占めています。 それはヘムオキシゲナーゼの主要な構成要素の一つである(Hooda et al. ら、2 0 1 4;Kurucz e t a l., 2018). オキシゲナーゼ−1の活性を増加させることは、老化する脳の酸化を遅延させることができる(Verdile et al. ら、2 0 1 5;SeriniおよびCalviello、2 0 1 6;Kuruczら、2 0 1 6;SeriniおよびCalviello、, 2018). これは広告に救済効果があります。 生理学的ストレスは、グルココルチコイドレベル上昇を誘導し、グルココルチコイドは、KLF4を介してヘムキャリアプロテイン1(HCP1)発現を増加させ、次いで、HCP1は、ヘムの取り込みを促進する(Li H.et a l., 2017). グルココルチコイドおよびKLF4は、共に抗炎症遺伝子を調節し、グルココルチコイド含量の低い細胞は、KLF4発現を完全に誘導することができない(Sevilla e t a l., 2015). ヘム摂取量のKLF4誘発性の増加は、脳内の鉄の蓄積につながります。 鉄は、ROSの放出を促進する(Tronel e t a l., 2013). 鉄の要素は心理的な圧力の下でラットの頭脳の酸化圧力を高めます(Yu et al., 2011). 従って、HCP1はKLF4およびglucocorticoidによって一緒に調整されるかもしれません。 Hcp1を増加させることは頭脳の鉄の蓄積に直接導き、酸化を悪化させ、apoptosisか機能障害を高め、そして頭脳の損傷を悪化させるヘムの通風管を高めます。
記憶障害と学習障害がADの主な症状であることは一般に認められています。 多数の臨床データは、ADにおける学習および認知機能障害の発症に対するA Βプラーク負荷および鉄蓄積応答が示されている(van Bergen e t a l., 2018). 最近発表されたデータは、高用量の鉄がA Β沈着を増加させ、マウスにおける学習および記憶を減衰させることを示唆している(Guo e t a l., 2013). 臨床研究では、鉄含有ミクログリアが磁気共鳴画像法下のAD患者の海馬に見出されることが示されている(Zeineh e t a l., 2015). ミクログリアは、転移または非転移、細胞外および細胞内供給源から鉄を獲得する(Mccarthy e t a l., 2018). 選択的かつ持続的なKLF4発現は、虚血性海馬反応性アストロサイトの核および細胞質において誘導することができる(Park e t a l., 2014). 研究では、KLF4が転写抑制剤として作用することが示されている。 それは、ELK−3の発現を下方調節し、次いで、ELK−3は、H O−1の発現を阻害する(Tsoyi e t a l., 2015). ヘムオキシゲナーゼ-1(HO-1)は、ヘムをビリルビン、遊離鉄、一酸化炭素に分解するストレスタンパク質である。 アストロサイトにおけるHO-1のアップレギュレーションは、脳内の異常な鉄沈着およびミトコンドリア機能障害をもたらし、認知能力の低下をもたら したがって、KLF4は、アストロサイトにおける鉄蓄積のプロセスに関与し、ADにおける酸化を悪化させ、脳損傷を悪化させる可能性がある。
結論
KLF4は、一般的に細胞増殖、アポトーシス、および分化を調節する上で極めて重要な役割を果たすことが知られている。 以前の研究は、神経炎症、神経保護およびシナプス再生を含むいくつかの重要な神経生理学的プロセスにおけるKLF4の調節に焦点を当てています。 最近、KLF4はADの病因において重要な役割を果たすことが見出されている。 この記事では、我々は神経保護とADにおける神経新生におけるKLF4の役割を確認します。
KLF4は、細胞の増殖と分化の調節の調節因子であるだけでなく、免疫応答を調節するための潜在的な標的でもある。 KLF4は、負の炎症因子を調節し、炎症反応を促進し、アストロサイト核ミクログリアの発現に大きな影響を与える可能性があります。 さらに、KLF4およびERK5は、神経保護作用を発揮するために一緒に作用することができる。 さらに、軸索再生は、特定の転写因子、細胞内阻害剤の含有量を変化させること、または細胞内シグナル伝達経路を変化させることによって達成するこ KLF4をたたくことは軸索の再生を高め、軸索の成長率を加速できます。 KLF4発現の減少は、JAK-STAT3経路を介して軸索再生を促進し、KLF4は、さらなる軸索再生へのJAK-STAT3経路を促進する。 したがって、KLF4は、抗炎症、抗アポトーシス、軸索再生とadの生成に重要な役割を果たしているCNSにおける鉄の蓄積のプロセスに関与している可能性が これらの知見は、KLF4は、ADのための潜在的な治療標的を表すことを示唆している。 しかし、KLF4のADへの影響の深い細胞および分子メカニズムは不明のままであり、さらなる調査が必要である。
著者の貢献
ZQC、XHZ、YJが原稿を書いた。 SHGとJYLは原稿の枠組みを変更しました。 BJLとRJCは重要な改訂を提供しました。 すべての著者は、提出のための原稿の最終版を承認しました。
資金調達
本研究は、中国自然科学財団(NSFC)(81871070、31571126、31471120)および吉林科技庁の資金調達(助成番号20180519003JH、20180414051GH、20170414034GHおよび20180414050
利益相反声明
著者らは、この研究は利益相反の可能性があると解釈される可能性のある商業的または財政的関係がない場合に行われたと宣言
Cacabelos,R.(2018). 過去5年間にアルツハイマー病の創薬に改善がありましたか。 経験値 オピン ドラッグディスコフ 13, 523–538. 土井: 10.1080/17460441.2018.1457645
PubMed Abstract|CrossRef全文|Google Scholar
ることができる。 マイクロアレイ解析による花粉シーズン中および外の季節性アレルギー性鼻炎の新規ターゲットの同定。 アクタ耳鼻咽喉科 135, 1330–1336. ドイ:10.3109/00016489.2015.1067906
PubMed要約/CrossRef全文/Google Scholar
ることができます。 アルツハイマー病におけるn-3PUFAの神経保護効果における脳内の酸化/窒素ストレスの減少およびその関与。 カー アルツハイマー病13,123-134. 土井: 10.2174/1567205012666150921101147
PubMed Abstract/CrossRef Full Text/Google Scholar