Dnaシークエンシングの技術は、Frederick Sangerのおかげで1977年に開発されました。 完全なゲノムを配列することが可能になるまでには少し時間がかかりました。 これは、より大きな完全なゲノムに数百万または数十億の小さな読み取りを組み立てるために、適切な数学モデルと大規模な計算能力が必要だった 今日の計算能力とソフトウェアは、2000年代初頭に何年もの作業を要していたものと、今日ではわずか数時間しかかからないものとの主な違いです。 あなたがこれをすることを選んだアルゴリズムは集まっている技術の”聖杯”である。 これらのアルゴリズムには、数学モデルで知られている最も有名な変数の1つであるk-merが組み込まれています。
k-merの起源とそれを取り巻く数学モデルは、数学関数の父として知られる1735年のスイスの数学者Leonhard Eulerに由来しています。 オランダの数学者Nicolaas de Bruijnは、オイラーのアイデアを適応させ、特定の長さのすべての可能な単語が循環シーケンス内の連続した文字列として正確に1回現れる、与えられたアルファベットから取られた文字の循環シーケンスを見つけました。
de Bruijnのアルゴリズムは分子生物学者によって適応され、何年も後にDNA配列をどのように組み立てるかという同等の問題に直面しました。 したがって、世界中の科学者は現在、De Bruijnグラフと変数kを使用しています。
DNA配列の組み立てへのk-mersの適用
いくつかの言葉では、de novoゲノムアセンブリは、連続した小さなDNA読み取りを接続し、より大きな配列で終わる De Bruijnグラフを生成するには(下の図を参照)、各読み取りのエッジのヌクレオチドが2番目のエッジ(など)と重なる必要があります。 最終的な目標は、連続した頂点を作成することであり、これは(潜在的に)大きなDNA断片になります。
あなたは、重複するヌクレオチドの特定の数であるk-mersにあなたの読み取りを断片化する必要があります。 K-merを使用すると、多くの小さなシーケンスから一意のシーケンスを生成することができます。 それぞれの一意のk-merシーケンスが識別され、余分なコピーが排除されます。 K-mersのこの側面は、次世代シーケンシングの欠点の一つを克服することができます—異なる周波数を持つゲノム領域を表す読み取りを取得する(すなわち、 K-mersの使用は不均等な順序の適用範囲のために複数回繰り返される順序を除去する。 しかし、k-merサイズが小さいほどヌクレオチドが重複する可能性が高くなり、値が大きいほどヌクレオチドが減少することに注意してください。
今日のde novoアセンブリ技術は、大きな読み取り(1.000–10.000bps)と小さな読み取り(100-200bps)のライブラリを使用すると、より効率的です。 ソフトウェア-プログラムはkの価値を使用でき、短い集まるのにk-mersは読みます。 これらはより正確なcontigsで終わるためにより大きい物によってそして組み込まれ、確認することができる。
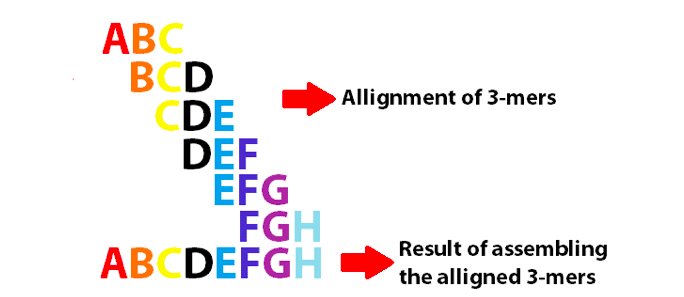
3-mersを使用して英語のアルファベットの8つの最初の文字を組み立てるde Bruijnグラフの例。 なお、これらの3-mersはk-1として重複する。
DNA Assembly
De Bruijnグラフをassemblyメソッドに適用し、最も適切なk-merサイズを選択する前に考慮する必要がある具体的なヒントがあります。 これらを活用することにより、より良い結果を生成できます。
- まず第一に、そしておそらく最も重要なのは、あなたのアセンブリで多くの異なるk-mersを使用することです。 その後、あなたの結果を評価し、最良のものを選択する必要があります(複数可)。 決して一つだけの正しいアセンブリはほとんど決してないことを忘れないでください。
- k-merを使用する前に、エラー読み取りを慎重に処理する必要があります。 慎重にエラーを削除しないと、結果が不要な膨らみを作成し、アセンブリが複雑になる可能性があります。 シーケンスのトリミング中に使用する誤り率のしきい値を増やします。 あなたはいくつかのシーケンスを失う可能性がありますが、残っている人は最高になります。
- 慎重に処理する必要があります。 例えば、Illumina sequencingは非常に大量のデータを生成します。 まず、読み取りのごく一部を組み立ててから、それらをすべて使用して違いを見つけてみてください。 反復可能な短い読み取りは、組立プロセスに悪影響を及ぼす可能性があります。
- 予想されるゲノムのサイズ、シークエンシングカバレッジの量、および読み取り数がわからない場合は、ゲノムの組み立てに最適なk値を選択する傾向があります。 Monash大学のvelvet advisorのようなk-mer advisorsを訪問して、どの値がより適しているかについてのアドバイスを得ることができます。
様々な長さのk-mersを使用し、連続体を整列させることは、研究者が突然変異率を発見するのにも役立ち、その使用を拡大する。 もちろん、組み立ての利益に向けてDe Bruijnグラフを操作することは万能薬ではありません。 生きている生物のゲノムを組み立てるための単純な機能よりも考慮すべき多くのことがあります。 これは単なる歴史の紹介であり、生物学者がそれをより効率的に使用する方法です。
- Compeau PE,Pevzner PA,Tesler G.(2011). ゲノムアセンブリへのde Bruijnグラフの適用方法。ネイチャー-バイオテクノロジー。 29(11):987–91.
- (2016). 拡張された配列コンテキストモデルは、ヒトゲノム全体の多型レベルの変動を広く説明する。 自然の遺伝学。 48(4): 349–55.

これはあなたを助けましたか? その後、あなたのネットワークと共有してくださ