introductie
het L1-celadhesiemolecuul (l1cam) is een neuronale celadhesiemolecuul dat behoort tot de immunoglobuline superfamilie; het bezit belangrijke functies in de ontwikkeling van het zenuwstelsel (Itoh en Fushiki, 2015). Mutaties in L1CAM zijn gerelateerd aan X-gebonden neurologische syndromen, die worden samengevat als L1 ziekten. Zij zijn als volgt ingedeeld:: X-linked hydrocephalus (XLH) als gevolg van stenose van het Aquaduct van Sylvius (HSAS), masa syndroom (intellectuele handicap, afasie, shuffling gang, geadducted thumbs), spastic paraparese type 1 (SP1), en X-linked agenesis van corpus callosum (ACC) (Weller and Gartner, 2001; Itoh and Fushiki, 2015).
ongeveer 282 ziekteverwekkende mutaties (DMS) in het l1cam-gen zijn gemeld in HGMD® Professional 2019,2 (https://portal.biobase-international.com/hgmd/pro/all.php). Veranderingen in het l1cam gen zijn gevarieerd; mutatiegegevens analyses van 282 patiënten onthullen 51% missense en nonsense mutaties, 25% deleties, 5% inserties en 19% splice site veranderingen, maar stille mutaties in L1CAM met pathogeen potentieel waren zeldzaam, en stille mutaties werden vaak genegeerd, vooral in whole-exome sequencing (WES) detectie.
in deze studie hebben we met WES het foetale DNA onderzocht van een Chinese zwangere vrouw die vijf continue zwangerschappen met foetale hydrocephalus heeft gemeld; we vonden alleen een nieuwe stille mutatie c.453G > T (p.Gly151 = ) in het l1cam-gen. Interessant genoeg, door verdere analyse, gaven we aan dat de Stille mutatie een potentiële 5′ splice site consensus sequentie creëerde, die zou resulteren in een in-frame verwijdering van 72 bp van exon 5 en 24 aminozuren van het l1cam eiwit.
Case Presentation
een 28-jarige gezonde vrouw werd naar onze kliniek verwezen na vier vrijwillige zwangerschapsonderbrekingen als gevolg van foetale hydrocefalie in andere ziekenhuizen. Alle foetussen waren mannelijk. Bij aankomst in ons ziekenhuis (Women ‘ s Hospital, School Of Medicine, Zhejiang University, Zhejiang, China), was ze al op haar vijfde zwangerschap op 24 weken zwangerschap, met een foetale hydrocephalus door beeldonderzoeken. Om de genetische oorzaak te onderzoeken, werd foetale bloedbemonstering uitgevoerd op 26 weken zwangerschapsleeftijd. Conventionele cytogenetische studies werden uitgevoerd voor zowel foetale als oudersteekproeven, en de foetale steekproef werd verder geanalyseerd door single-nucleotide polymorphism (SNP) array en WES.
deze studie werd uitgevoerd in overeenstemming met de aanbevelingen van de Ethische Commissie van het Women ‘ s Hospital, School Of Medicine Zhejiang University, en er werd geïnformeerde toestemming verkregen van alle deelnemers aan deze studie in overeenstemming met de Verklaring van Helsinki. Het studieprotocol werd goedgekeurd door de beoordelingscommissie van het Women ‘ s Hospital, School Of Medicine, Zhejiang University in China.
materialen en methoden
Karyotype en SNP Array
de karyotypen van foetaal navelstrengbloed en perifeer navelstrengbloed werden bepaald door conventionele karyotypering van ten minste 30 bloedlymfocyten, die bij metafase door colchicines werden gearresteerd. G-banding karyotypes van gekweekte cellen werden uitgevoerd op het niveau van de 320-400-band met een resolutie van rond 10 Mb. SNP array werd uitgevoerd door de CytoScan™ HD array (Affymetrix, USA) volgens de instructies van de fabrikant, met ongeveer 2.600.000 markers, waaronder 750.000 SNP sondes en 1.900.000 niet-polymorfisme sondes voor uitgebreide gehele genoom dekking. De gegevens werden geanalyseerd door de chromosoomanalyse Suite (ChAS) software (Affymetrix, Santa Clara, CA) gebaseerd op de grch37/hg19 assemblage. De rapportagedrempel voor het resultaat van het kopieernummer werd vastgesteld op 500 kb met een markergetal van ≥50 voor winsten en op 200 kb met een markergetal van ≥50 voor verliezen.
gehele Exoomsequencing
het grootste deel van WES werd geleverd door het Beijing Genomics Institute. Genomisch DNA werd geëxtraheerd door een Dneasy Bloedkit (Qiagen, CA) en vervolgens gefragmenteerd door Covaris LE220 (Massachusetts, USA) om een gepaarde-eindbibliotheek (200-250 bp) te produceren. Alle versterkte bibliotheken werden uitgevoerd op het bgiseq-500 platform, werd het enig-bundel DNA gemengd met MGIEasy™ DNA-bibliotheek Prep Kit V1 (BGI, Shenzhen, China) en vervolgens gerangschikt gebruikend 100SR chemie met bgiseq-500RS high-throughput sequencing Kit (BGI, Shenzhen, China).
Clean reads (met een lengte van 90 bp) afgeleid van gerichte sequencing en filtering werden vervolgens uitgelijnd op de human genome reference (hg19) met behulp van het Burrows-Wheeler Aligner (BWA) Multi-Vision softwarepakket (Li and Durbin, 2009). Na uitlijning, werden de output dossiers gebruikt om het rangschikken dekking en diepteanalyse van het doelgebied uit te voeren, single-nucleotide varianten (SNVs), en Indel roepen, gebruikten we de GATK software om SNVs en indels te ontdekken (McKenna et al., 2010), werden alle SNV ‘ s en indels gefilterd en geschat via meerdere databases, waaronder de National Center for Biotechnology Information (NCBI) Single-Nucleotide Polymorphism Database (dbSNP), HapMap, 1000 Genomen Project dataset, en database van 100 Chinese gezonde volwassenen. We gebruikten Condel, zift, Polyfen-2, LRT, Mutatieproever en PhyloP om het effect van varianten te voorspellen. Pathogene varianten worden beoordeeld op grond van het protocol uitgegeven door het American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015). De database van de menselijke genmutatie (Hgmd) werd gebruikt om mutaties te screenen. Alle potentiële pathogene varianten werden gevalideerd met behulp van Sanger sequencing methoden.
RNA-extractie, PCR en Sequencing
mononucleaire cellen uit perifeer bloed (Pmbc ‘s) en mononucleaire cellen uit navelstrengbloed (CBMC’ s) werden geïsoleerd door scheiding van de Ficoll-dichtheid. Totaal RNA werd geëxtraheerd uit Pmbc ’s en CBMC’ s met behulp van TRIzol (Takara, Japan). Geëxtraheerde totale RNAs werden reverse-getranscribeerd met behulp van RT Kit (Takara, Japan). PCR werd uitgevoerd met behulp van GoldStar beste Master Mix (Cwbio, Beijing). Primer sequenties zijn vermeld: L1CAM-DNA – 5F, CCCACCCGTCCTTTCCTA; L1CAM-DNA-5R, CGCTCGTCCTTGATGT; L1CAM-mRNA-4-6-F, GGTGTCCACTTCAAACCCAA; en L1CAM-mRNA-4-6-R, GCGGCTTCCTGTCAATCA. Het rangschikken van Sanger werd uitgevoerd door een ABI 3130 de Analysator van DNA.
resultaten
een gezonde vrouw van 28 jaar werd naar onze kliniek verwezen na vier vrijwillige zwangerschapsonderbrekingen als gevolg van foetale hydrocephalus. Alle foetussen waren mannelijk (figuur 1A). De familiale stamboom leek XLH te laten zien. Ze was hier al op de vijfde zwangerschap na 26 weken zwangerschap. Foetale ventriculomegalie werd gedetecteerd door foetale echografie en MRI, die consequent de aanwezigheid van hydrocephalus aangetoond. Ze toonden aan dat de bilaterale cerebrale ventrikel en de derde ventrikel duidelijk verwijd waren, en er was ernstige hydrocephalus in de intracerebrale en agenese van het corpus callosum (figuur 1B).
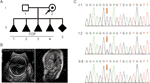
figuur 1 (A) stamboom van het gezin. TOP, beëindiging van de zwangerschap. B) beeldvormingsonderzoeken van de foetus. Foetale echografie en foetale MRI toonde aan dat er ernstige hydrocephalus in de foetus. C) Sequentieanalyse van genomisch DNA van familieleden. De genotypes van L1CAM waren wild type, c. 453G > T Het, en c. 453G > T Hom, in I:1 (echtgenoot), i:2 (zwangere vrouw), en II:5 (foetus). De mutatie wordt aangegeven door de rode pijlen.
om de mogelijke genetische oorzaak te onderzoeken, voerden we karyotype analyse en SNP array uit om de foetale bloedmonsters te analyseren en vonden we geen positieve bevindingen. Choroïdale neovascularisatie (CNV) is gedeponeerd in de genexpressie Omnibus (GEO); het toetredingsnummer is GSE133063, zoals hieronder vermeld (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
toen ontdekten we de foetus door WES. De analytische strategie voor het vinden van waarschijnlijke pathogene variant identificatie werd getoond in Figuur S1. Een lijst van varianten (tabel S1) werd verkregen door de screening van variantfrequenties, mutatiestatus en overerving modus. Rekening houdend met de hydrocephalus-geassocieerde genen (HP:0000238, http://compbio.charite.de/hpoweb/showterm?id=HP:0000238#id=HP_0000238) (tabel S2), was er geen bijkomende opmerkelijke mutatie behalve de Stille mutatie van c.453G > T in exon 5 van het l1cam-gen (NM_000425.3). C. 453G > T werd niet gerapporteerd in HGMD en ClinVar en werd niet gevonden in dbSNP, gnomAD en andere datasets. Volgens de normen en richtlijnen van de ACMG (Richards et al., 2015), had het nog niet het criterium van “pathogeen” of “waarschijnlijk pathogeen” bereikt, maar er waren geen andere potentiële mutaties; we hadden geen andere keuze dan een verdere analyse van de gevonden stille mutatie te maken.
volgens het traditionele denken vond deze basissubstitutie plaats in de derde base in codon 151, dat codeert voor een glycine, waardoor een neutrale mutatie ontstaat (p.Gly151 = ). Deze variant werd bevestigd in DNA uit foetaal navelstrengbloed en perifeer bloed in het paar door het rangschikken van Sanger (figuur 1C). De vrouw droeg de heterozygote mutatie, en haar man was een wild-type genotype.
met Mutatieproever (http://www.mutationtaster.org/), c.453G > T werd gescoord als “ziekte veroorzakend.”Het toonde aan dat eiwit functies kunnen worden beïnvloed en de splice site kan worden veranderd; we waren benieuwd naar de potentiële splicing effecten van de l1cam-functie in deze stille mutatie. De Stille mutatie werd getest met behulp van de volgende online softwareproducten: NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) en NNSplice (http://www.fruitfly.org/seq_tools/splice.html); de 5′ potentiële splice-site werd ook voorspeld te worden gecreëerd in de L1CAM c.453G > t mutatie met behulp van de softwareproducten (figuur S2). De resultaten toonden aan dat deze stille mutatie een potentiële 5′ splice site 72 bp stroomopwaarts van de normale exon 6/intron 6 splice site creëerde (figuur 2A). Als dit het geval is, kunnen we de lengteverandering van l1cam messenger RNA (mRNA) vinden tussen I:2 en II:5 (Figuur 2A). RT-PCR werd uitgevoerd gebruikend primers ontworpen om exons 4-6 in l1cam mRNA te versterken. Inderdaad, de resultaten toonden een korte band van afkaping in foetale cDNA PCR (II: 5), terwijl de band versterkt van l1cam mRNA bevatte de verwachte lange band in man cDNA PCR (I:1) en lange / korte bands bij de zwangere vrouw cDNA PCR (i: 2) (Figuur 2B). Het directe rangschikken van het versterkte fragment toonde aan dat de schrapping laatste 72 bp van exon 5 in mannelijke foetale cDNA impliceerde (de vrouw was een drager) (figuur 2C). We hebben het cruciale pathogene bewijs.
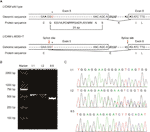
Figuur 2 (A) schematische weergave van exon 5, intron 6 en exon 6 organisatie in L1CAM. (B) RT-PCR analyse van exonen 5 en 6 van de l1cam cDNA uit mononucleaire cellen uit perifeer bloed (Pmbc ‘s) en mononucleaire cellen uit navelstrengbloed (CBMC’ s). Agarosegelelektroforese van RT-PCR-producten gegenereerd uit I:1 (man), i:2 (zwangere vrouw), en II: 5 (foetus). C) Sequentieanalyse van het RT-PCR-product uit Pmbc ’s van het paar en CBMC’ s van de foetus.
deze stille mutatie resulteerde in 24 aminozuren van l1cam-eiwit (residuen 151-174); Lys (K) werd vervangen door Glu (E) bij codon 175 (figuur 2A). Er waren uitlijning van meerdere l1cam eiwitsequenties over verschillende soorten en behoud van de ontbrekende aminozuren in l1cam over zoogdieren: Homo sapiens, Pan troglodytes, Bos taurus, Mus musculus en Rattus norvegicus (figuur 3A). Wild-type en c.453G > T splicing mutatie l1cam eiwitten werden voorspeld door de software Cphmodels-3.2 Server (http://www.cbs.dtu.dk/services/CPHmodels/) (figuur 3B). Immunoglobuline-achtig (IG-achtig) domein 2 (residuen 134-230) van wildtype-en splicingmutatie l1cam-eiwitten is weergegeven in Figuur 3C. L1CAM c.453G > T splicingmutatie veranderde de eiwitstructuur, vooral het IG-achtig domein 2

Figuur 3 (A) uitlijning van meerdere l1cam-eiwitsequenties over verschillende soorten. De L1CAM c.453G > t resulteerde in 24 aminozuren van l1cam-eiwit (residuen 151-174) die ontbraken in het geconserveerde aminozuurgebied bij verschillende soorten. De zwarte kolom toont de ontbrekende aminozuren. B) de structuren van wild-type en c.453G > T splicing mutatie L1CAM eiwit zoals voorspeld door de software Cphmodels-3.2 Server. C) de structuren van immunoglobuline-achtig (IG-achtig) domein 2 (residuen 134-230) van wild-type en splicing-mutatie L1CAM-eiwit.
discussie
Stille mutaties werden vaak gedetecteerd door WES, maar er werd onvoldoende aandacht besteed, wat leidde tot het weglaten van DMs. In deze studie, hebben we WES gebruikt om de genetische oorzaak van een Chinese familie met hydrocefalie te onderzoeken, maar vonden alleen een nieuwe stille mutatie in L1CAM, die ons dwong om een verdere analyse te maken. Gelukkig hebben we bewezen dat de Stille mutatie een nieuwe 5′ splice site creëerde en een DM was.
mutaties in L1CAM kunnen een X-gebonden L1-ziekte veroorzaken, maar de klinische symptomen zijn variabel; mutaties produceren onverwachte fenotypen. In de studie zijn de vijf lijdende foetussen allemaal mannetjes, wat overeenkomt met een overerving patroon. De foetale echografie en MRI tonen een typische L1 ziekte, met inbegrip van XLH en agenesis van het corpus callosum. Het verbetert ons inzicht in de genotype-fenotype correlatie van L1CAM.
L1CAM c. 453G > T (p. Gly151 =) werd aanvankelijk verondersteld geen effect te hebben op de eiwitsequentie. Maar andere stille mutaties, c.924C > T (p.Gly308 = ) en C.645C > T (p.Gly215 = ), in het l1cam gen zijn gemeld als DMs (Du et al., 1998; Vos et al., 2010). De c.924C > T mutatie resulteerde in de activering van een nieuwe splitsingsplaats 69 bp 5 ‘op de normale splitsingsplaats exon 8/intron 8 donor, en het is verklaard als een” ziekteveroorzakende ” plaats voor hydrocefalie (Du et al., 1998). Voor c.645C > T in L1CAM werd 51 bp verwijderd met de activering van een nieuwe exon 6/intron 6 donor splice (Vos et al., 2010). Onze huidige studie was vergelijkbaar; de mutatie van ca. 453G > t creëerde een potentiële 5’ splice site stroomopwaarts van de normale exon 5/intron 5 splice site. Al deze stille mutaties creëerden nieuwe donor splice sites, resulterend in de exon overgeslagen. Het herinnerde ons eraan om veel aandacht te besteden aan deze stille mutaties, die het verbinden van eiwitten kunnen beïnvloeden.
als transmembraanglycoproteïne en lid van de superfamilie van immunoglobuline van celadhesiemoleculen kan het l1cam-eiwit aan het celoppervlak interageren met een aantal verschillende glycoproteïnen, en homofiele binding is waarschijnlijk de belangrijkste interactie (Wei and Ryu, 2012). De studies naar de kristalstructuur van IG-achtige domeinen 1-4 in neurofascin suggereerden dat veel pathologische L1 mutaties behouden aminozuurresiduen binnen deze domeinen beïnvloeden en interfereren met homofiele interacties (Liu et al., 2011), vooral zoals geverifieerd door de functie onderzoek van IG-achtige domein 2 (Zhao et al., 1998). In onze studie speculeerden we dat L1CAM c.453G > T IG-achtig domein 2 veranderde in het extracellulaire deel van het l1cam-eiwit, wat leidde tot de abnormale extracellulaire interactie, waarbij het niet lukte om stroomafwaarts de signaalroute te starten. Een verdere studie gewijd aan de massaspectrometrie van deze l1cam variant zou specifiek verduidelijken welk moleculair ensemble in de cel wordt geproduceerd.
samenvattend rapporteerden we via WES een nieuwe stille mutatie c. 453G > T in L1CAM die een 5′ splice site produceert die verantwoordelijk is voor hydrocefalie. Deze abnormale eiwitvariant zou Ig-achtig domein 2 veranderen, wat invloed zou kunnen hebben op de homofiele binding van het l1cam-eiwit. Daarnaast hebben we prenatale genetische diagnostiek uitgevoerd voor de zwangere vrouw die vijf continue zwangerschappen met hydrocefalie meldde. Ondertussen stelde het voor dat sommige stille mutaties die in WES worden ontdekt niet zouden moeten worden genegeerd; splicing voorspellingen van deze mutaties waren noodzakelijk. Het leverde een nieuwe genetische basis voor prenatale diagnose en pre-implantatie prenatale diagnose van hydrocephalus.
beschikbaarheid van gegevens
openbaar beschikbare datasets werden geanalyseerd in deze studie. Deze gegevens zijn hier te vinden: GSE133063 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
ethische verklaring
de studies met menselijke deelnemers werden beoordeeld en goedgekeurd door de beoordelingscommissie van het Women ‘ s Hospital, School Of Medicine, Zhejiang University in China. Schriftelijke geà nformeerde toestemming om deel te nemen aan deze studie werd verstrekt door de wettelijke voogd/nabestaanden van de deelnemers. Schriftelijke geà nformeerde toestemming werd verkregen van de persoon(s), en de wettelijke voogd(en) van de minderjarige (s)/nabestaanden, voor de publicatie van mogelijk identificeerbare afbeeldingen of gegevens die in dit artikel zijn opgenomen.
Auteursbijdragen
ys, YLi, MC, YLu, YQ en YY voerden experimenten uit. YS heeft de cijfers opgesteld. MC en YLi hebben de WES data geanalyseerd. YLu en YQ verrichtten karyotype analyse en SNP array. Je hebt monsters gerekruteerd. HL en FL verstrekten beeldvormingsonderzoeken. YS en MD schreven het manuscript. Alle auteurs hebben het definitieve manuscript gelezen en goedgekeurd.
financiering
deze studie werd gesteund door de National Natural Science Foundation of China (subsidie nr. 81801441), het belangrijkste onderzoeks-en ontwikkelingsprogramma van de provincie Zhejiang (subsidie Nr. 81801441). 2019C03025), het nationale belangrijke onderzoeks-en ontwikkelingsprogramma van China (subsidie Nr. 2016YFC1000703), en de medische wetenschappelijke Onderzoeksstichting van de provincie Zhejiang (subsidie Nr. 2014KYA246).
verklaring inzake belangenconflicten
de auteurs verklaren dat het onderzoek werd uitgevoerd zonder enige commerciële of financiële relatie die als een potentieel belangenconflict kon worden opgevat.
erkenningen
we danken de patiënten die deelnamen aan dit onderzoek. Wij danken Dr. Jiong Gao (BGI Genomics, BGI-Shenzhen, Shenzhen 518083, China) voor zijn hulp bij de voorbereiding van dit manuscript.
aanvullend materiaal
het aanvullende materiaal voor dit artikel is online te vinden op: https://www.frontiersin.org/articles/10.3389/fgene.2019.00817/full#supplementary-material
figuur S1 / analytische strategie voor het vinden van waarschijnlijke pathogene variant identificatie door WES.
figuur S2 / Donorsplitsingsplaatsen voorspeld door NetGene2 en NNSplice.
tabel S1 / lijst van varianten door het screenen van varianten frequenties, mutatiestatus en overerving modus.
tabel S2 / lijst van Hydrocefalie-geassocieerde genen (Export voor HP:0000238).
Du, Y. Z., Dickerson, C., Aylsworth, A. S., Schwartz, C. E. (1998). Een stille mutatie, C924T (G308G), in het l1cam gen resulteert in X-linked hydrocephalus (HSA ‘ s). J. Med. Genet. 35 (6), 456–462. doi: 10.1136 / jmg.35.6.456
PubMed Abstract / CrossRef Full Text / Google Scholar
Itoh, K., Fushiki, S. (2015). De rol van L1cam in muriene corticogenese, en de pathogenese van hydrocephalus. Pathol. Int. 65 (2), 58–66. doi: 10.1111 / pin.12245
PubMed Abstract / CrossRef Full Text / Google Scholar
Li, H., Durbin, R. (2009). Snelle en nauwkeurige korte uitlijning met Burrows-Wheeler transformator. Bioinformatica 25 (14), 1754-1760. doi: 10.1093 / bioinformatics / btp324
PubMed Abstract / CrossRef Full Text / Google Scholar
Liu, H., Focia, P. J., He, X. (2011). Homofiel adhesiemechanisme van neurofascin, een lid van de L1-familie van neurale celadhesiemoleculen. J. Biol. Scheikunde. 286 (1), 797–805. doi: 10.1074 / jbc.M110.180281
PubMed Abstract / CrossRef Full Text / Google Scholar
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). De Genome Analysis Toolkit: een MapReduce framework voor het analyseren van de volgende generatie DNA sequencing data. Genome Res. 20 (9), 1297-1303. doi: 10.1101 / gr.107524.110
PubMed Abstract / CrossRef Full Text / Google Scholar
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standaarden en richtlijnen voor de interpretatie van sequentievarianten: een gezamenlijke consensusaanbeveling van het American College of Medical Genetics and Genomics en de Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi: 10.1038 / gim.2015.30
PubMed Abstract / CrossRef Full Text / Google Scholar
Vos, Y. J., De Walle, H. E. K., Bos, K. K., Stegeman, J. A., Berge, A. M., Bruining, M., et al. (2010). Genotype-fenotype correlaties bij L1 syndroom: een gids voor genetische counselling en mutatieanalyse. J. Med. Genet. 47 (3), 169–175. doi: 10.1136 / jmg.2009.071688
PubMed Abstract / CrossRef Full Text / Google Scholar
Wei, C. H., Ryu, S. E. (2012). Homofiele interactie van de L1 familie van de molecules van de celadhesie. Exp. Mol. Med. 44 (7), 413–423. doi: 10.3858 / emm.2012.44.7.050
PubMed Abstract / CrossRef Full Text / Google Scholar
Weller, S., Gartner, J. (2001). Genetische en klinische aspecten van X-linked hydrocephalus( L1 ziekte): mutaties in het l1cam gen. Brom. Mutat. 18 (1), 1–12. doi: 10.1002 / humu.1144
PubMed Abstract | CrossRef Full Text | Google Scholar
Zhao, X., Yip, P. M., Siu, C. H. (1998). Identification of a homophilic binding site in immunoglobulin-like domain 2 of the cell adhesion molecule L1. J. Neurochem. 71 (3), 960–971 doi: 10.1046/j.1471-4159.1998.71030960.x.
PubMed Abstract | CrossRef Full Text | Google Scholar