de technologie voor het rangschikken van DNA werd ontwikkeld terug in 1977 dankzij Frederick Sanger. Het duurde wat langer voordat het mogelijk was om een volledig genoom te sequencen. Dit komt omdat we een geschikt wiskundig model en een enorme rekenkracht nodig hadden om miljoenen of miljarden kleine reads te assembleren tot een groter volledig genoom. De huidige computationele kracht en software zijn het belangrijkste verschil tussen wat vroeger jaren werk in de vroege jaren 2000 en wat duurt slechts een paar uur vandaag. Het algoritme dat je koos om dit te doen is de “Heilige Graal” van de assemblage technologie. Deze algoritmen bevatten een van de beroemdste variabelen die bekend zijn in wiskundige modellen, de k-mer.
de oorsprong van de k-mer en het mathematische model dat eromheen ligt, is afkomstig van de Zwitserse wiskundige Leonhard Euler uit 1735, die bekend staat als de vader van de wiskundige functie. Een Nederlandse wiskundige Nicolaas de Bruijn paste Eulers ideeën aan om een cyclische reeks letters uit een bepaald alfabet te vinden waarvoor elk woord van een bepaalde lengte precies één keer als een reeks opeenvolgende tekens in de cyclische reeks voorkomt.
het algoritme van de Bruijn werd aangepast door moleculair biologen, die vele jaren later geconfronteerd werden met een soortgelijk probleem: hoe DNA-sequenties te assembleren. Zo gebruiken wetenschappers over de hele wereld nu de grafiek De Bruijn en de variabele k.
toepassing van k-mers op het samenstellen van DNA-sequenties
In enkele woorden, de novo genoom assemblage impliceert het verbinden van opeenvolgende kleine DNA-reads en het eindigen met grotere sequenties. Om een De Bruijn-grafiek te genereren (zie onderstaande figuur), moeten de nucleotiden aan de rand van elke lezing de rand van een tweede overlappen (enzovoort). Het uiteindelijke doel is om een opeenvolgende vertex te creëren, die (potentieel) zal resulteren in grote DNA-fragmenten.
u moet uw reads fragmenteren in k-mers, een specifiek aantal nucleotiden die elkaar overlappen. Met de k-mer kunt u een unieke sequentie genereren uit vele kleine. Elke unieke K-mer sequentie wordt geïdentificeerd en extra kopieën worden geëlimineerd. Dit aspect van k-mers staat u toe om één van de nadelen van volgende generatie het rangschikken te overwinnen — het krijgen leest die genomic gebieden met verschillende frequenties vertegenwoordigen (d.w.z., het krijgen van heel wat kleine leest van één gebied). Het gebruik van k-mers elimineert opeenvolgingen die meer dan eens wegens ongelijke opeenvolgingsdekking worden herhaald. Nochtans, houd in gedachten dat een lage K-mer grootte de kansen voor nucleotiden het overlappen zal verhogen, terwijl het hebben van een grotere waarde hen zal verminderen.
de huidige De novo-assemblagetechnologie is efficiënter wanneer u gebruik maakt van bibliotheken met grote leeseenheden (d.w.z. 1.000–10.000 bps) gecombineerd met kleinere bibliotheken (100-200 bps). Softwareprogramma ‘ s kunnen de K-waarde en k-mers gebruiken om korte reads te assembleren. Deze kunnen dan worden opgenomen en geverifieerd door grotere degenen om te eindigen in nauwkeuriger contigs.
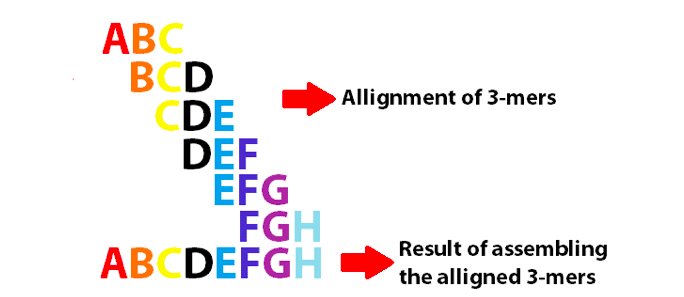
voorbeeld van een de Bruijn grafiek met behulp van 3-mers om de eerste 8 letters van het Engelse alfabet samen te stellen. Merk op dat deze 3-mers overlappen als k-1.
hoe meer u weet hoe meer U kunt bereiken in DNA-assemblage
er zijn specifieke tips die u moet overwegen voordat u de Bruijn-grafieken in uw assemblagemethode toepast en de meest geschikte k-mer-maat kiest. Door deze te benutten, kunt u betere resultaten te genereren.
- Allereerst, en misschien wel het belangrijkste, is het gebruik van veel verschillende k-mers in uw assemblage. U moet dan evalueren uw resultaten en kies de beste(s). Vergeet nooit dat er bijna nooit één en slechts één correcte assemblage is.
- u moet zorgvuldig omgaan met foutlezingen, voordat u een k-mer gebruikt. Als u de fouten niet zorgvuldig verwijdert, kunnen de resultaten een ongewenste bobbel creëren, waardoor uw assemblage wordt bemoeilijkt. Verhoog de drempelwaarde voor het foutenpercentage dat u gebruikt tijdens het bijsnijden van sequenties. Je kunt een aantal sequenties verliezen, maar degenen die overblijven zullen de beste zijn.
- u moet zorgvuldig omgaan met DNA-herhalingen. Bijvoorbeeld, genereert het rangschikken Illumina een zeer grote hoeveelheid gegevens. Probeer eerst een klein deel van de reads samen te stellen en gebruik ze allemaal om verschillen te spotten. Herhaalbare korte reads kunnen negatief interfereren met uw assemblageproces.
- Ken uw gegevens. Als u de grootte van uw verwachte genoom, de hoeveelheid het rangschikken dekking, en het aantal leest niet kent, dan bent u meer geneigd om de beste K-waarde voor het assembleren van uw genoom te kiezen. U kunt k-mer adviseurs bezoeken, zoals velvet advisor van Monash university om advies te krijgen over welke waarde geschikter lijkt.
het gebruik van k-mers van verschillende lengtes en het uitlijnen van de contigs helpt onderzoekers ook mutatiepercentages te herkennen, waardoor het gebruik ervan wordt uitgebreid. Natuurlijk is het manipuleren van de Bruijn grafieken naar assemblagevoordelen geen wondermiddel. Er zijn tal van dingen te overwegen dan een simplistische functie voor het samenstellen van het genoom van een levend organisme. Dit is slechts een introductie van de geschiedenis en hoe biologen het efficiënter kunnen gebruiken.
- Compeau PE, Pevzner PA, Tesler G. (2011). Hoe de Bruijn grafieken toe te passen op genoomassemblage.Natuur Biotechnologie. 29(11):987–91.
- Aggarwala V, Voight BF. (2016). Een uitgebreid sequentiecontextmodel verklaart breed de variabiliteit in polymorfismeniveaus in het menselijke genoom. Natuurgenetica. 48(4): 349–55.

heeft dit u geholpen? Dan kunt u delen met uw netwerk.