- Introdução
- Apresentação Do Caso
- Materiais e Métodos
- Cariótipo e SNP Array
- sequenciamento de Exoma inteiro
- extração de RNA, PCR e sequenciamento
- resultados
- discussão
- disponibilidade de dados
- Declaração de Ética
- contribuições do autor
- financiamento
- Declaração de conflito de interesses
- agradecimentos
- material suplementar
Introdução
L1 célula, molécula de adesão gene (L1CAM) é uma molécula de adesão celular neuronal pertencentes à imunoglobulina superfamily; possui funções-chave no desenvolvimento do sistema nervoso (Itoh e Fushiki, 2015). Mutações em L1CAM têm sido relacionadas a síndromes neurológicas ligadas ao X, que são resumidas como doenças L1. Eles são classificados da seguinte forma: X-linked hidrocefalia (XLH) devido a estenose do aqueduto de Sylvius (HSAS), MASA (síndrome de deficiência mental, afasia, misturando marcha, adducted polegares), paraparesia espástica do tipo 1 (SP1), e X-vinculada a agenesia do corpo caloso (ACC) (Weller e Gartner, 2001; Itoh e Fushiki, 2015).Cerca de 282 mutações causadoras de doenças (DMs) no gene L1CAM foram relatadas no Hgmd® Professional 2019.2 (https://portal.biobase-international.com/hgmd/pro/all.php). Alterações no gene L1CAM são variadas; as análises de dados de mutação de 282 pacientes revelam 51% de missense e mutações sem sentido, 25% de deleções, 5% de inserções e 19% de alterações no local de emenda, mas mutações silenciosas em L1CAM com potencial patogênico eram raras, e mutações silenciosas eram frequentemente ignoradas especialmente na detecção de sequenciamento de Exoma inteiro (WES).
neste estudo, usando WES, examinamos o DNA fetal de uma mulher grávida chinesa que relatou cinco gestações contínuas com hidrocefalia fetal; encontramos apenas uma nova mutação silenciosa C. 453g > T (P.Gly151 = ) no gene L1CAM. Curiosamente, por meio de análises adicionais, indicamos que a mutação silenciosa criou uma potencial sequência de consenso do local de emenda de 5′, o que resultaria em uma exclusão em quadro de 72 bp do exon 5 e 24 aminoácidos da proteína L1CAM.
Apresentação Do Caso
uma mulher saudável de 28 anos foi encaminhada para nossa clínica após quatro terminações voluntárias de gravidez devido a hidrocefalia fetal em outros hospitais. Todos os fetos eram do sexo masculino. Ao chegar ao nosso hospital (Hospital Feminino, Escola de Medicina, Universidade de Zhejiang, Zhejiang, China), ela já estava na quinta gravidez às 24 semanas de gestação, com hidrocefalia fetal por exames de imagem. Para explorar a causa genética, a amostragem de sangue fetal foi realizada às 26 semanas de idade gestacional. Estudos citogenéticos convencionais foram realizados para amostras fetais e parentais, e a amostra fetal foi ainda analisada por matriz de polimorfismo de nucleotídeo único (SNP) e Wes.
este estudo foi realizado de acordo com as recomendações do Comitê de Ética do Hospital da Mulher, Escola de Medicina da Universidade de Zhejiang, e o consentimento informado foi adquirido de todos os participantes deste estudo de acordo com a Declaração de Helsinque. O protocolo do estudo foi aprovado pelo Conselho de revisão do Hospital da Mulher, Escola de Medicina, Universidade de Zhejiang na China.
Materiais e Métodos
Cariótipo e SNP Array
O cariótipo fetal de sangue de cordão umbilical e periférica de sangue de cordão umbilical foi determinada através da avaliação do cariótipo convencional de, pelo menos, 30 linfócitos de sangue, que foram presos em metafase por colchicina. Cariótipos de bandas G de células cultivadas foram realizados no nível de 320-400 bandas com uma resolução de cerca de 10 Mb. A matriz SNP foi realizada pelo cytoscan ™ HD array (Affymetrix, EUA) de acordo com as instruções do fabricante, com cerca de 2.600.000 marcadores, incluindo 750.000 sondas SNP e 1.900.000 sondas não polimorfismo para cobertura abrangente do genoma inteiro. Os dados foram analisados pelo software cromossomo Analysis Suite (ChAS) (Affymetrix, Santa Clara, CA) com base na montagem GRCh37/hg19. O limiar de reporte do resultado do número de cópia foi fixado em 500 kb com uma contagem de marcadores ≥50 para ganhos e em 200 kb com uma contagem de marcadores ≥50 para perdas.
sequenciamento de Exoma inteiro
A parte principal do WES foi fornecida pelo Instituto de genômica de Pequim. O DNA genômico foi extraído por um kit de sangue DNeasy (Qiagen, CA) e então foi fragmentado por Covaris LE220 (Massachusetts, EUA) para gerar uma biblioteca de extremidade pareada (200-250 bp). Todas as bibliotecas amplificadas foram realizadas na plataforma BGISEQ-500, o DNA de fita única foi misturado com MGIEasy™ DNA Library Prep Kit V1 (BGI, Shenzhen, China) e depois sequenciado usando 100SR química com Bgiseq-500Rs high-throughput sequencing Kit (BGI, Shenzhen, China).
leituras limpas (com um comprimento de 90 PB) derivadas de sequenciamento e filtragem direcionados foram então alinhadas à referência do genoma humano (hg19) usando o pacote de software Multi-visão Burrows-Wheeler Aligner (BWA) (Li e Durbin, 2009). Após o alinhamento, os arquivos de saída foram usados para realizar a cobertura de sequenciamento e análise de profundidade da região alvo, variantes de nucleotídeo único (SNVs) e chamada indel, usamos o software GATK para detectar SNVs e indels (McKenna et al., 2010), Todos os SNVs e indels foram filtrados e estimados por meio de vários bancos de dados, incluindo o National Center for Biotechnology Information (NCBI) Single-Nucleotide Polymorphism Database (dbSNP), HapMap, 1000 Genomes Project dataset e banco de dados de 100 adultos saudáveis chineses. Usamos Condel, SIFT, PolyPhen-2, LRT, Mutation Taster e PhyloP para prever o efeito das variantes. As variantes patogênicas são avaliadas sob o protocolo emitido pelo American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015). O Human gene Mutation Database (HGMD) foi usado para rastrear mutações. Todas as variantes patogênicas potenciais foram validadas usando métodos de sequenciamento Sanger.
extração de RNA, PCR e sequenciamento
células mononucleares do sangue periférico (PMBCs) e células mononucleares do sangue do cordão umbilical (CBMCs) foram isoladas por separação de gradiente de densidade de Ficoll. O RNA Total foi extraído do PMBCs e do CBMCs usando TRIzol (Takara, Japão). Os RNAs totais extraídos foram transcritos reversamente usando Kit RT (Takara, Japão). A PCR foi realizada usando GoldStar Best Master Mix (Cwbio, Pequim). As sequências de Primer estão listadas: L1CAM-DNA-5F, CCCACCGTCCTTCCTA; L1CAM-DNA-5R, CGCTCGTCCTGCTTGATGT; L1cam-mRNA-4-6-F, GGTGTCCACTTCAAACCAA; e l1cam-mRNA-4-6-R, GCGGCTTCCTGTCAATCA. O sequenciamento de Sanger foi realizado por um analisador de DNA ABI 3130.
resultados
uma mulher saudável de 28 anos foi encaminhada para nossa clínica após quatro terminações voluntárias de gravidez devido a hidrocefalia fetal. Todos os fetos eram do sexo masculino (figura 1a). O pedigree familiar parecia mostrar XLH. Ela já estava aqui quinta gravidez às 26 semanas de gestação. A ventriculomegalia Fetal foi detectada por ultrassonografia fetal e ressonância magnética, o que demonstrou consistentemente a presença de hidrocefalia. Eles mostraram que o ventrículo cerebral bilateral e o terceiro ventrículo estavam obviamente dilatados, e havia hidrocefalia grave na intracerebral e na agenesia do corpo caloso (figura 1b).
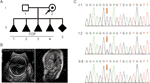
Figura 1 (A) Pedigree da família. Topo, interrupção da gravidez. (B) exames de imagem do feto. A ultrassonografia Fetal e a ressonância magnética fetal mostraram que havia hidrocefalia grave no feto. (C) Análise de sequência de DNA genômico de membros da família. Os genótipos de L1CAM foram do tipo selvagem, C. 453g > T Het, e C. 453g > T Hom, em I:1 (marido), I: 2 (mulher grávida) e II: 5 (feto). A mutação é indicada pelas setas vermelhas.
para explorar a possível causa genética, realizamos análise cariotípica e matriz SNP para analisar a amostragem de sangue fetal e não encontramos achados positivos. O resultado da neovascularização coroidal (CNV) foi depositado na expressão gênica Omnibus (GEO); o número de adesão é GSE133063, conforme anexado abaixo (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
então, detectamos o feto por WES. A estratégia analítica para encontrar provável identificação de variante patogênica foi mostrada na figura S1. Uma lista de variantes (tabela S1) foi obtida através da triagem de frequências variantes, status de mutação e modo de herança. Levando em consideração os genes associados à hidrocefalia (HP:0000238, http://compbio.charite.de/hpoweb/showterm?id=HP:0000238#id=HP_0000238) (tabela S2), não houve mutação notável adicional, exceto a mutação silenciosa de C. 453g > T no exon 5 do gene L1CAM (NM_000425.3). C. 453g > T não foi relatado em HGMD e ClinVar e não foi encontrado em dbSNP, gnomAD e outros conjuntos de dados. De acordo com os padrões e diretrizes da ACMG (Richards et al., 2015), ainda não havia atingido o critério de “patogênico” ou “provavelmente patogênico”, mas não havia outras mutações potenciais; não tivemos escolha a não ser fazer uma análise mais aprofundada da mutação silenciosa encontrada.
de Acordo com o pensamento tradicional, esta base de substituição ocorreu na terceira base no codão 151, que codifica uma glicina, criando, assim, uma mutação neutra (p.Gly151 = ). Esta variante foi confirmada no DNA extraído do sangue do cordão umbilical fetal e sangue periférico no casal por sequenciamento de Sanger (figura 1C). A mulher carregava a mutação heterozigótica, e seu marido era um genótipo de tipo selvagem.
com provador de mutação (http://www.mutationtaster.org/), C. 453g > T foi classificado como ” causador de doenças.”Mostrou que as características da proteína puderam ser afetadas e o local da tala pôde ser mudado; nós estávamos curiosos sobre os efeitos de emenda potenciais da função de L1CAM nesta mutação silenciosa. A mutação silenciosa foi testada usando os seguintes produtos de software online: NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) e NNSplice (http://www.fruitfly.org/seq_tools/splice.html); o site de emenda potencial 5′ também foi previsto para ser criado no L1CAM C.453g > mutação t usando os produtos de software (figura S2). Os resultados mostraram que esta mutação silenciosa criou um potencial 5 ‘ splice site 72 bp a montante do exon normal 6 / intron 6 splice site (figura 2a). Se este for o caso, podemos encontrar a mudança de comprimento do RNA mensageiro L1CAM (mRNA) entre I:2 e II:5 (Figura 2A). A RT-PCR foi realizada usando primers projetados para amplificar exons 4-6 no mRNA L1CAM. De fato, os resultados mostraram uma banda curta de truncamento no PCR de cDNA fetal (II:5), enquanto a banda amplificada do mRNA L1CAM continha a banda longa esperada no PCR de cDNA do marido (I:1) e bandas longas / curtas na gestante cDNA PCR (i: 2) (Figura 2B). O sequenciamento direto do fragmento amplificado mostrou que a deleção envolveu os últimos 72 Pb do exon 5 no cDNA fetal masculino (a mulher era portadora) (figura 2C). Temos as evidências patogênicas cruciais.
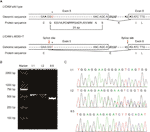
a Figura 2 (A) representação Esquemática do éxon 5, intron 6, e exão 6 organização em L1CAM. (B) análise RT-PCR de exons 5 e 6 do cDNA L1CAM de células mononucleares do sangue periférico (PMBCs) e células mononucleares do sangue do cordão umbilical (CBMCs). Eletroforese em gel de Agarose de produtos RT-PCR gerados a partir de I:1 (marido), I:2 (mulher grávida) e II:5 (feto). (C) Análise de sequência do produto RT-PCR de PMBCs do casal e CBMCs do feto.
esta mutação silenciosa resultou em 24 aminoácidos da proteína L1CAM (resíduos 151-174); Lys (K) foi substituído por Glu (E) no códon 175 (figura 2a). Houve alinhamento de várias sequências de proteínas L1CAM em várias espécies e conservação dos aminoácidos ausentes em l1cam em mamíferos: Homo sapiens, Pan troglodytes, Bos taurus, Mus musculus e Rattus norvegicus (figura 3A). Wild-type E C. 453g > t splicing mutation as proteínas L1CAM foram previstas pelo software Cphmodels-3.2 Server (http://www.cbs.dtu.dk/services/CPHmodels/) (figura 3b). Immunoglobulin-like (Ig-like) domínio 2 (resíduos 134-230) de tipo selvagem e emenda de mutação L1CAM proteínas é mostrado na Figura 3C. L1CAM c.453G > T splicing mutação alterada a estrutura da proteína, especialmente Ig-como domínio 2

a Figura 3 (A) Alinhamento de várias L1CAM de seqüências de proteína em todas as espécies. O L1CAM C.453G > t resultou em 24 aminoácidos da proteína L1CAM (resíduos 151-174) ausentes na região de aminoácidos conservada em diferentes espécies. A coluna preta mostra os aminoácidos ausentes. (B) as estruturas de tipo selvagem e C. 453g > t splicing mutação l1cam proteína conforme previsto pelo software Cphmodels-3.2 Server. (C) As estruturas de imunoglobulina-like (Ig-like) domínio 2 (resíduos 134-230) de tipo selvagem e splicing mutação proteína L1CAM.
discussão
mutações silenciosas foram frequentemente detectadas pelo WES, mas atenção insuficiente foi dada, levando à omissão de DMs. Neste estudo, empregamos o WES para explorar a causa genética de uma família chinesa com hidrocefalia, mas só encontramos uma nova mutação silenciosa no L1CAM, que nos forçou a fazer uma análise mais aprofundada. Felizmente, provamos que a mutação silenciosa criou um novo local de emenda de 5′ e era um DM.Mutações em L1cam podem causar uma doença L1 ligada ao X, mas os sintomas clínicos são variáveis; mutações produzem fenótipos inesperados. No estudo, os cinco fetos sofredores são todos do sexo masculino, o que é consistente com um padrão de herança. A ultrassonografia fetal e a ressonância magnética mostram uma doença L1 típica, incluindo XLH e agenesia do corpo caloso. Melhora nossa compreensão sobre a correlação genótipo–fenótipo de L1CAM.
L1CAM C. 453g > T (P.Gly151 = ) foi inicialmente pensado para não ter efeito sobre a sequência de proteínas. Mas outras mutações silenciosas, C. 924c > T (P.Gly308 = ) e C. 645c > T (P. Gly215 = ), no gene L1CAM foram relatadas como DMs (Du et al., 1998; Vos et al., 2010). C.A mutação 924C > t resultou na ativação de um novo local de emenda 69 bp 5 ‘para o local normal de emenda do doador exon 8/intron 8, e foi declarado como um local” causador de doenças ” para hidrocefalia (Du et al., 1998). Para C. 645C > T em L1CAM, 51 PB foi excluído com a ativação de uma nova emenda de doador exon 6 / intron 6 (Vos et al., 2010). Nosso presente estudo foi semelhante; a mutação de C. 453g > t criou um potencial local de emenda de 5’ a montante do exon normal 5/intron 5 local de emenda. Todas essas mutações silenciosas criaram novos locais de emenda de doadores, resultando no exon sendo ignorado. Isso nos lembrou de prestar muita atenção a essas mutações silenciosas, que podem afetar a emenda de proteínas.
como glicoproteína transmembrana e membro da superfamília de imunoglobulinas das moléculas de adesão celular, a proteína L1CAM pode interagir na superfície celular com várias glicoproteínas diferentes, e a ligação homofílica é provavelmente seu principal modo de interação (Wei e Ryu, 2012). Os estudos sobre a estrutura cristalina dos domínios semelhantes ao Ig 1-4 na neurofascina sugeriram que muitas mutações patológicas L1 afetam os resíduos de aminoácidos conservados nesses domínios e interferem nas interações homofílicas (Liu et al., 2011), especialmente conforme verificado pela função Pesquisa do domínio IG-like 2 (Zhao et al., 1998). Em nosso estudo, especulamos que L1CAM C. 453g > t alterou o domínio IG-like 2 na parte extracelular da proteína L1CAM, levando à interação extracelular anormal, não conseguindo iniciar a jusante da via de sinalização. Um estudo adicional dedicado à espectrometria de massa desta variante L1CAM esclareceria especificamente qual conjunto molecular é produzido na célula.
em resumo, por meio do WES, relatamos uma nova mutação silenciosa C. 453g > T em L1CAM que produz um local de emenda de 5′ responsável pela hidrocefalia. Esta variante anormal da proteína foi prevista para alterar o domínio Ig-like 2, o que pode afetar a ligação homofílica da proteína l1cam. Além disso, realizamos diagnóstico genético pré-natal para a gestante relatando cinco gestações contínuas com hidrocefalia. Enquanto isso, sugeriu que algumas mutações silenciosas detectadas no WES não deveriam ser ignoradas; as previsões de emenda dessas mutações eram necessárias. Forneceu uma nova base genética para o diagnóstico pré-natal e diagnóstico pré-natal pré-implantação de hidrocefalia.
disponibilidade de dados
conjuntos de dados disponíveis publicamente foram analisados neste estudo. Estes dados podem ser encontrados aqui: GSE133063(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
Declaração de Ética
os estudos envolvendo participantes humanos foram revisados e aprovados pelo Conselho de revisão do Hospital da Mulher, Escola de Medicina, Universidade de Zhejiang, na China. O consentimento informado por escrito para participar deste estudo foi fornecido pelo tutor legal/parente mais próximo dos participantes. O consentimento informado por escrito foi obtido do(s) indivíduo (s) e do (s) tutor (es) legal (s) do menor (s), para a publicação de quaisquer imagens ou dados potencialmente identificáveis incluídos neste artigo.
contribuições do autor
Ys, YLi, MC, YLu, YQ e YY realizaram experimentos. Ys preparou os números. MC e YLi analisaram os dados do WES. YLu e YQ realizaram análise de cariótipo e matriz SNP. YY recrutou amostras. HL e FL forneceram exames de imagem. YS e MD escreveram o manuscrito. Todos os autores leram e aprovaram o manuscrito final.
financiamento
este estudo foi apoiado pela Fundação Nacional de Ciências Naturais da China (Grant No. 81801441), o principal programa de pesquisa e desenvolvimento da província de Zhejiang (Grant No. 2019c03025), o Programa Chave Nacional da investigação e desenvolvimento de China (concessão No. 2016yfc1000703), e a Fundação de investigação científica médica da província de Zhejiang (concessão No. 2014kya246).
Declaração de conflito de interesses
os autores declaram que a pesquisa foi realizada na ausência de quaisquer relações comerciais ou financeiras que pudessem ser interpretadas como um potencial conflito de interesses.
agradecimentos
agradecemos aos pacientes inscritos nesta pesquisa. Agradecemos ao Dr. Jiong Gao (BGI Genomics, BGI-Shenzhen, Shenzhen 518083, China) por sua assistência durante a preparação deste manuscrito.
material suplementar
o material suplementar para este artigo pode ser encontrado online em: https://www.frontiersin.org/articles/10.3389/fgene.2019.00817/full#supplementary-material
figura S1 / estratégia analítica para encontrar provável identificação de variante patogênica pelo WES.
figura S2 / locais de emenda de doadores previstos por NetGene2 e NNSplice.
tabela S1 / lista de variantes por meio da triagem de frequências de variantes, status de mutação e modo de herança.
tabela S2 / lista de genes associados à hidrocefalia (exportação para HP:0000238).
Du, Y. Z., Dickerson, C., Aylsworth, A. S., Schwartz, C. E. (1998). Uma mutação silenciosa, C924T (G308G), no gene L1CAM resulta em hidrocefalia ligada ao X (HSAS). J. Med. Genet. 35 (6), 456–462. doi: 10.1136 / jmg.35.6.456
PubMed Abstract / CrossRef Texto Completo / Google Scholar
Itoh, K., Fushiki, S. (2015). O papel da L1cam na corticogênese murina e na patogênese da hidrocefalia. Pathol. T. 65 (2), 58–66. doi: 10.1111 / pin.12245
PubMed Abstract / CrossRef Texto Completo / Google Scholar
Li, H., Durbin, R. (2009). Alinhamento rápido e preciso de Leitura curta com Burrows-Wheeler transform. Bioinformática 25 (14), 1754-1760. doi: 10.1093/Bioinformática / btp324
resumo PubMed / CrossRef texto completo / Google Scholar
Liu, H., foco, P. J., He, X. (2011). Mecanismo de adesão homofílica da neurofascina, um membro da família L1 de moléculas de adesão de células neurais. J. Biol. Chem. 286 (1), 797–805. doi: 10.1074 / jbc.M110.180281
PubMed Abstract / CrossRef texto completo / Google Scholar
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). O Genome Analysis Toolkit: uma estrutura MapReduce para analisar dados de sequenciamento de DNA de próxima geração. Genoma Res. 20 (9), 1297-1303. doi: 10.1101 / gr.107524.110
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Ferreira, J., et al. (2015). Padrões e diretrizes para a interpretação de variantes de sequência: uma recomendação de consenso conjunta do American College of Medical Genetics and Genomics e da Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi: 10.1038 / gim.2015.30
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Vos, Y. J., de Walle, H. E. K., Bos, K. K., Stegeman, J. A., Berge, A. M., Bruining, M., et al. (2010). Correlações genótipo-fenótipo na síndrome L1: um guia para aconselhamento genético e análise de mutação. J. Med. Genet. 47 (3), 169–175. doi: 10.1136 / jmg.2009.071688
PubMed Abstract / CrossRef Texto Completo / Google Scholar
Wei, C. H., Ryu, S. E. (2012). Interação homofílica da família L1 de moléculas de adesão celular. Expo. Mol. Med. 44 (7), 413–423. doi: 10.3858 / emm.2012.44.7.050
PubMed Abstract / CrossRef Texto Completo / Google Scholar
Weller, S., Gartner, J. (2001). Aspectos genéticos e clínicos da hidrocefalia ligada ao X( doença L1): mutações no gene L1CAM. Cantarolar. Mutat. 18 (1), 1–12. doi: 10.1002 / humu.1144
PubMed Abstract | CrossRef Full Text | Google Scholar
Zhao, X., Yip, P. M., Siu, C. H. (1998). Identification of a homophilic binding site in immunoglobulin-like domain 2 of the cell adhesion molecule L1. J. Neurochem. 71 (3), 960–971 doi: 10.1046/j.1471-4159.1998.71030960.x.
PubMed Abstract | CrossRef Full Text | Google Scholar