a tecnologia para sequenciamento de DNA foi desenvolvida em 1977 graças a Frederick Sanger. Demorou um pouco mais para que fosse possível sequenciar um genoma completo. Isso ocorre porque precisávamos de um modelo matemático apropriado e de um enorme poder computacional para montar milhões ou bilhões de pequenas leituras em um genoma completo maior. O poder computacional e o software de hoje são a principal diferença entre o que costumava levar anos de trabalho no início dos anos 2000 e o que leva apenas algumas horas hoje. O algoritmo que você escolheu para fazer isso é o” Santo Graal ” da tecnologia de montagem. Esses algoritmos incorporam uma das variáveis mais famosas conhecidas em modelos matemáticos, o k-mer.A origem do k-mer e do modelo matemático que o rodeia vem de um matemático suíço de 1735 Leonhard Euler, que é conhecido como o pai da função matemática. Um matemático holandês Nicolaas De Bruijn adaptou as idéias de Euler para encontrar uma sequência cíclica de letras tiradas de um determinado alfabeto para o qual cada palavra possível de um certo comprimento aparece como uma sequência de caracteres consecutivos na sequência cíclica exatamente uma vez.O algoritmo de Bruijn foi adaptado por biólogos moleculares, que muitos anos depois enfrentaram um problema equivalente: como montar sequências de DNA. Assim, cientistas de todo o mundo agora usam o gráfico de Bruijn e a variável K.
aplicação de K-mers à montagem de sequências de DNA
em poucas palavras, a montagem do genoma de novo envolve a conexão de pequenas leituras consecutivas de DNA e terminando com sequências maiores. Para gerar um gráfico de Bruijn (veja a figura abaixo), os nucleotídeos na borda de cada leitura Devem se sobrepor à borda de um segundo (e assim por diante). O objetivo final é criar um vértice consecutivo, que (potencialmente) resultará em grandes fragmentos de DNA.
você tem que fragmentar suas leituras em K-mers, que são um número específico de nucleotídeos que se sobrepõem. O k-mer permite gerar uma sequência única de muitos pequenos. Cada sequência k-mer única é identificada e cópias extras são eliminadas. Esse aspecto do k-mers permite superar uma das desvantagens do sequenciamento de próxima geração — obter leituras que representam regiões genômicas com frequências diferentes (ou seja, obter muitas leituras pequenas de uma região). O uso de K-mers elimina sequências repetidas mais de uma vez devido à cobertura desigual da sequência. No entanto, tenha em mente que um tamanho baixo de K-mer aumentará as chances de sobreposição de nucleotídeos, enquanto ter um valor maior os diminuirá.
a tecnologia de montagem de novo de hoje é mais eficiente quando você usa bibliotecas de leituras grandes (ou seja, 1.000–10.000 bps) combinadas com outras menores (100-200 bps). Os programas de Software podem usar o valor k E k-mers para montar leituras curtas. Estes podem então ser incorporados e verificados por outros maiores para acabar em contigs mais precisos.
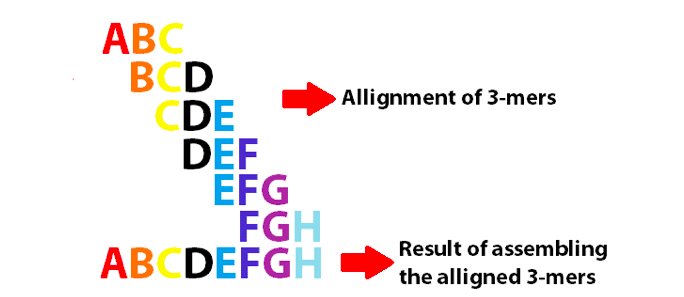
exemplo de um gráfico de Bruijn usando 3-mers para montar as 8 primeiras letras do alfabeto Inglês. Observe que esses 3-mers se sobrepõem como k-1.
quanto mais Você sabe, mais Você pode conseguir na montagem de DNA
existem dicas específicas que você precisa considerar antes de aplicar Gráficos De Bruijn em seu método de montagem e escolher o tamanho K-mer mais apropriado. Ao aproveitar estes, você pode gerar melhores resultados.
- em primeiro lugar, e talvez o mais importante, é usar muitos K-mers diferentes em sua montagem. Você deve então avaliar seus resultados e escolher o(s) melhor (s). Nunca se esqueça que quase nunca há uma e apenas uma montagem correta.
- você deve lidar cuidadosamente com leituras de erro, antes de usar um K-mer. Se você não remover cuidadosamente os erros, os resultados podem criar uma protuberância indesejada, complicando sua montagem. Aumente o limite para a taxa de erro que você usa durante o corte de sequência. Você pode perder algumas sequências, mas aqueles que permanecem serão os melhores.
- você deve lidar com repetições de DNA cuidadosamente. Por exemplo, o sequenciamento Illumina gera uma quantidade muito grande de dados. Primeiro, tente montar uma pequena fração das leituras e, em seguida, use-as todas para detectar diferenças. Leituras curtas repetíveis podem interferir negativamente no seu processo de montagem.
- Conheça seus dados. Se você não sabe o tamanho do seu genoma esperado, a quantidade de cobertura de sequenciamento e o número de leituras, então você está mais propenso a escolher o melhor valor k para montar seu genoma. Você pode visitar K-Mer advisors, como o Velvet advisor da Monash university para obter alguns conselhos sobre qual valor parece mais adequado.
usando K-mers de vários comprimentos e alinhando os contigs também ajuda os pesquisadores a detectar taxas de mutação, expandindo seu uso. Claro, manipular gráficos de Bruijn para benefício de montagem não é uma panacéia. Existem inúmeras coisas a considerar do que uma função simplista para montar o genoma de um organismo vivo. Esta é apenas uma introdução da história e como os biólogos podem usá-la com mais eficiência.
- Compeau PE, Pevzner PA, Tesler G. (2011). Como aplicar Gráficos De Bruijn à montagem do genoma.Biotecnologia Da Natureza. 29(11):987–91.
- Aggarwala V, Voight BF. (2016). Um modelo de contexto de sequência expandida explica amplamente a variabilidade nos níveis de polimorfismo em todo o genoma humano. Genética Da Natureza. 48(4): 349–55.

isso ajudou você? Então, por favor, compartilhe com sua rede.