tehnologia pentru secvențierea ADN a fost dezvoltată în 1977 datorită lui Frederick Sanger. A durat un pic mai mult înainte de a fi posibilă secvența unui genom complet. Acest lucru se datorează faptului că aveam nevoie de un model matematic adecvat și de o putere de calcul masivă pentru a asambla milioane sau miliarde de citiri mici într-un genom complet mai mare. Puterea computațională de astăzi și software-ul sunt principala diferență între ceea ce obișnuia să dureze ani de muncă la începutul anilor 2000 și ceea ce durează doar câteva ore astăzi. Algoritmul pe care l-ați ales pentru a face acest lucru este „sfântul graal” al tehnologiei de asamblare. Acești algoritmi încorporează una dintre cele mai cunoscute variabile cunoscute în modelele matematice, k-mer.
originea k-mer și modelul matematic care îl înconjoară provine de la un matematician elvețian din 1735 Leonhard Euler, care este cunoscut ca tatăl funcției matematice. Un matematician olandez Nicolaas de Bruijn a adaptat ideile lui Euler pentru a găsi o secvență ciclică de Litere preluate dintr-un alfabet dat pentru care fiecare cuvânt posibil de o anumită lungime apare ca un șir de caractere consecutive în secvența ciclică exact o dată.
algoritmul lui De Bruijn a fost adaptat de biologi moleculari, care mulți ani mai târziu s-au confruntat cu o problemă echivalentă: cum se asamblează secvențele ADN. Astfel, oamenii de știință din întreaga lume folosesc acum graficul de Bruijn și variabila k.
aplicarea k-mers la asamblarea secvențelor ADN
în câteva cuvinte, asamblarea genomului de novo implică conectarea citirilor consecutive ale ADN-ului mic și încheierea cu secvențe mai mari. Pentru a genera un grafic de Bruijn (vezi figura de mai jos), nucleotidele de la marginea fiecărei citiri trebuie să se suprapună cu marginea unei a doua (și așa mai departe). Scopul final este de a crea un vârf consecutiv, care (potențial) va duce la fragmente mari de ADN.
trebuie să fragmentați citirile în k-mers, care sunt un număr specific de nucleotide care se suprapun. K-mer vă permite să generați o secvență unică de la multe mici. Fiecare secvență unică k-mer este identificată și copiile suplimentare sunt eliminate. Acest aspect al K-mers vă permite să depășiți unul dintre dezavantajele secvențierii de generație următoare — obținerea de lecturi care reprezintă regiuni genomice cu frecvențe diferite (adică obținerea multor lecturi mici dintr-o regiune). Utilizarea k-mers elimină secvențele repetate de mai multe ori din cauza acoperirii inegale a secvenței. Cu toate acestea, rețineți că o dimensiune scăzută a k-mer va crește șansele de suprapunere a nucleotidelor, în timp ce o valoare mai mare le va scădea.
tehnologia de asamblare de novo de astăzi este mai eficientă atunci când utilizați biblioteci de citiri mari (adică 1.000–10.000 bps) combinate cu cele mai mici (100-200 bps). Programele Software pot utiliza valoarea k și K-mers pentru a asambla citiri scurte. Acestea pot fi apoi încorporate și verificate de cele mai mari pentru a ajunge în contiguri mai precise.
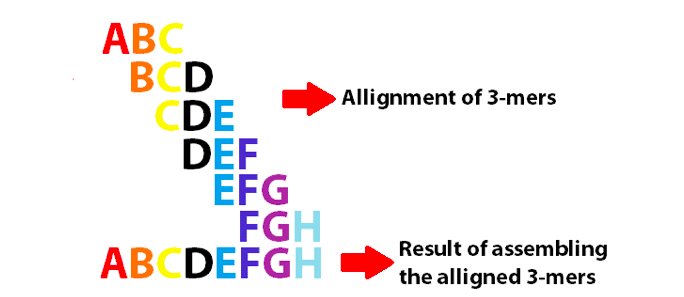
exemplu de grafic de Bruijn folosind 3-mers pentru a asambla primele 8 litere ale alfabetului englez. Rețineți că aceste 3-mers se suprapun ca k-1.
cu cât știți mai mult, cu atât puteți obține mai mult în asamblarea ADN
există sfaturi specifice pe care trebuie să le luați în considerare înainte de a aplica graficele de Bruijn în metoda dvs. de asamblare și de a alege cea mai potrivită dimensiune k-mer. Prin valorificarea acestora, puteți genera rezultate mai bune.
- în primul rând, și poate cel mai important, este să folosiți multe k-mers diferite în adunarea voastră. Apoi, ar trebui să vă evaluați rezultatele și să alegeți cel mai bun(E). Nu uitați niciodată că nu există aproape niciodată un singur ansamblu corect.
- ar trebui să se ocupe cu atenție erori citește, înainte de a utiliza un k-mer. Dacă nu eliminați cu atenție Erorile, rezultatele pot crea o umflătură nedorită, complicând asamblarea. Măriți pragul pentru rata de eroare pe care o utilizați în timpul tăierii secvenței. S-ar putea pierde unele secvențe, dar cei care rămân vor fi cei mai buni.
- ar trebui să se ocupe cu atenție ADN-ul se repetă. De exemplu, secvențierea Illumina generează o cantitate foarte mare de date. În primul rând, încercați să asamblați o mică parte din citiri, apoi folosiți-le pe toate pentru a observa diferențele. Citirile scurte repetabile pot interfera negativ cu procesul de asamblare.
- cunoaște-ți datele. Dacă nu cunoașteți dimensiunea genomului așteptat, cantitatea de acoperire a secvențierii și numărul de citiri, atunci sunteți mai predispus să alegeți cea mai bună valoare k pentru asamblarea genomului. Puteți vizita k-mer advisors, cum ar fi velvet advisor de la Universitatea Monash pentru a obține câteva sfaturi despre ce valoare pare mai potrivită.
utilizarea k-mers de diferite lungimi și alinierea contigilor ajută, de asemenea, cercetătorii să identifice ratele de mutație, extinzând utilizarea acesteia. Desigur, manipularea graficelor de Bruijn către beneficiul asamblării nu este un panaceu. Există numeroase lucruri de luat în considerare decât o funcție simplistă pentru asamblarea genomului unui organism viu. Aceasta este doar o introducere a istoriei și a modului în care biologii o pot folosi mai eficient.
- Compeau PE, Pevzner PA, Tesler G. (2011). Cum se aplică graficele de Bruijn la asamblarea genomului.Biotehnologia Naturii. 29(11):987–91.
- Aggarwala V, Voight BF. (2016). Un model de context de secvență extins explică în linii mari variabilitatea nivelurilor de polimorfism din genomul uman. Genetica Naturii. 48(4): 349–55.

v-a ajutat acest lucru? Apoi, vă rugăm să partajați cu rețeaua.