technologia sekwencjonowania DNA została opracowana w 1977 roku dzięki Frederickowi Sangerowi. Zajęło to trochę więcej czasu, zanim było możliwe zsekwencjonowanie pełnego genomu. To dlatego, że potrzebowaliśmy odpowiedniego modelu matematycznego i ogromnej mocy obliczeniowej, aby zebrać miliony lub miliardy małych odczytów do większego kompletnego genomu. Dzisiejsza moc obliczeniowa i oprogramowanie są główną różnicą między tym, co kiedyś zajmowało lata pracy na początku 2000 roku, a tym, co dziś zajmuje tylko kilka godzin. Algorytm, który wybrałeś, to „Święty Graal” technologii montażu. Algorytmy te zawierają jedną z najbardziej znanych zmiennych znanych w modelach matematycznych, k-mer.
pochodzenie k-mer i modelu matematycznego, który go otacza, pochodzi od szwajcarskiego matematyka Leonharda Eulera z 1735 roku, który jest znany jako ojciec funkcji matematycznej. Holenderski matematyk Nicolaas de Bruijn zaadaptował idee Eulera, aby znaleźć cykliczny ciąg liter z danego alfabetu, dla którego każde możliwe słowo o określonej długości pojawia się jako ciąg kolejnych znaków w sekwencji cyklicznej dokładnie raz.
algorytm de Bruijna został zaadaptowany przez biologów molekularnych, którzy wiele lat później stanęli przed równorzędnym problemem: jak zmontować sekwencje DNA. Tak więc naukowcy na całym świecie używają teraz grafu de Bruijna i zmiennej K.
zastosowanie k-merów Do Montażu sekwencji DNA
w kilku słowach, de novo montaż genomu polega na połączeniu kolejnych małych odczytów DNA i zakończeniu większych sekwencji. Aby wygenerować wykres de Bruijna (patrz rysunek poniżej), nukleotydy na krawędzi każdego odczytu muszą pokrywać się z krawędzią drugiego (i tak dalej). Ostatecznym celem jest stworzenie kolejnych wierzchołków, co (potencjalnie) spowoduje powstanie dużych fragmentów DNA.
musisz podzielić swoje odczyty na k-Mery, które są określoną liczbą nukleotydów, które nakładają się na siebie. K-mer pozwala na wygenerowanie unikalnej sekwencji z wielu małych. Każda unikalna Sekwencja k-mer jest identyfikowana, a dodatkowe kopie są eliminowane. Ten aspekt k-mers pozwala przezwyciężyć jedną z wad sekwencjonowania następnej generacji-uzyskanie odczytów, które reprezentują regiony genomowe o różnych częstotliwościach (tj. uzyskanie wielu małych odczytów z jednego regionu). Zastosowanie k-mers eliminuje sekwencje powtarzane więcej niż jeden raz z powodu nierównego pokrycia sekwencji. Należy jednak pamiętać, że niski rozmiar k-mer zwiększy szanse na nakładanie się nukleotydów, podczas gdy posiadanie większej wartości zmniejszy je.
dzisiejsza technologia montażu de novo jest bardziej wydajna, gdy używa się bibliotek o dużych odczytach (np. 1.000–10.000 bps) w połączeniu z mniejszymi (100-200 bps). Programy mogą używać wartości k I k-mers do montażu krótkich odczytów. Można je następnie włączyć i zweryfikować przez większe, aby uzyskać dokładniejsze kontrasty.
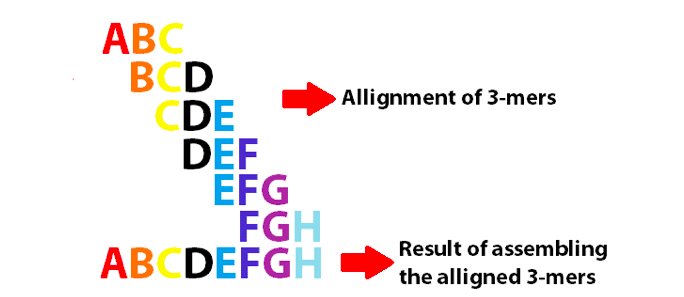
przykład grafu de Bruijna wykorzystującego 3-mers do złożenia 8 pierwszych liter alfabetu angielskiego. Zauważ, że te 3-Mery pokrywają się jako k-1.
im więcej wiesz, tym więcej możesz osiągnąć w montażu DNA
istnieją konkretne wskazówki, które należy Rozważyć Przed zastosowaniem Wykresów De Bruijn w metodzie montażu i wyborem najbardziej odpowiedniego rozmiaru k-mer. Wykorzystując je, można generować lepsze wyniki.
- po pierwsze, a może najważniejsze, jest użycie wielu różnych k-mers w swoim montażu. Następnie należy ocenić wyniki i wybrać najlepsze z nich. Nigdy nie zapominaj, że prawie nigdy nie ma jednego i tylko jednego poprawnego montażu.
- przed użyciem k-mer należy ostrożnie obchodzić się z odczytami błędów. Jeśli nie usuniesz ostrożnie błędów, wyniki mogą spowodować niechciane wybrzuszenie, komplikując montaż. Zwiększ próg błędu używanego podczas przycinania sekwencji. Możesz stracić kilka sekwencji, ale ci, którzy pozostaną, będą najlepsi.
- należy ostrożnie obchodzić się z powtórzeniami DNA. Na przykład sekwencjonowanie Illumina generuje bardzo dużą ilość danych. Najpierw spróbuj zebrać niewielką część czytań, a następnie użyj ich wszystkich, aby dostrzec różnice. Powtarzalne krótkie odczyty mogą negatywnie wpływać na proces montażu.
- Poznaj swoje dane. Jeśli nie znasz wielkości oczekiwanego genomu, ilości sekwencjonowania pokrycia i liczby odczytów, to jesteś bardziej skłonny do wyboru najlepszej wartości k do montażu genomu. Możesz odwiedzić doradców k-mer, takich jak velvet advisor z Monash university, aby uzyskać porady, która wartość wydaje się bardziej odpowiednia.
używanie k-merów o różnych długościach i wyrównywanie stygów pomaga również badaczom wykryć wskaźniki mutacji, rozszerzając ich zastosowanie. Oczywiście manipulowanie wykresami De Bruijna w kierunku korzyści montażowych nie jest panaceum. Jest wiele rzeczy do rozważenia niż uproszczona funkcja montażu genomu żywego organizmu. To tylko wprowadzenie do historii i tego, jak biolodzy mogą ją efektywniej wykorzystać.
- Compeau PE, Pevzner PA, Tesler G. (2011). Jak zastosować wykresy De Bruijn do montażu genomu.Biotechnologia Przyrody. 29(11):987–91.
- Aggarwala V, Voight BF. (2016). Rozszerzony model kontekstu sekwencji szeroko wyjaśnia zmienność poziomów polimorfizmu w całym ludzkim genomie. Genetyka Natury. 48(4): 349–55.

czy to ci pomogło? Następnie podziel się z siecią.