wprowadzenie
kruppel-like factor 4 (KLF4) jest członkiem rodziny zinc-finger transcription factor,która jest wyrażana w różnych tkankach ludzkich. Jest dobrze znany jako jeden z czterech czynników indukcji do pluripotencjalnych komórek macierzystych (iPSCs) (Ghaleb and Yang, 2017). KLF4 może regulować wiele ważnych procesów biologicznych, takich jak neuroinflammation, stres oksydacyjny, proliferacja, różnicowanie i apoptoza (Kaushik et al., 2010; Mamonkin et al., 2013; Zhang et al., 2015; Miao et al., 2017; Xu et al., 2017). Ilości poprzednich badań koncentrowały się na roli KLF4 w rozwoju i progresji raka (Karam et al., 2017; Yadav et al., 2018). KLF4 jest dwufunkcyjnym czynnikiem transkrypcyjnym, który może pełnić swoją rolę jako onkogen lub Gen supresorowy nowotworu w zależności od rodzaju raka lub stadium raka (Evans and Liu, 2008). Może aktywować lub hamować transkrypcję genów zaangażowanych w proliferację, różnicowanie i apoptozę komórek (Ding i in., 2015). KLF4 może współpracować z innymi czynnikami przeprogramowującymi, aby przekształcić komórki somatyczne w iPSCs i hamować różnicowanie komórek macierzystych (Takahashi and Yamanaka, 2006; van Schaijik et al., 2018). Zapewnia to perspektywy terapeutyczne dla chorób naczyniowych, chorób immunologicznych, anoreksji i innych chorób (Imbernon et al., 2014; Liu Y. et al., 2015; Murgai et al., 2017). Ponadto KLF4 może odgrywać szeroko regulacyjną rolę w ośrodkowym układzie nerwowym (OUN). Kilka badań wskazuje, że KLF4 jest związany z wieloma zaburzeniami neurologicznymi, w tym chorobą Alzheimera (AD), epilepsją, chorobą Parkinsona, wodogłowiem i schizofrenią (Qin et al., 2011; Xie et al., 2013; Han et al., 2015; Nishiguchi et al., 2015; Li L. et al., 2017).
AD jest jedną z najczęstszych przewlekłych chorób neurodegeneracyjnych, która prowadzi do upośledzenia funkcji poznawczych i pamięci, różnych objawów psychicznych i zaburzeń behawioralnych, a postępująca demencja jest najczęstszą cechą kliniczną (Jiang et al., 2018). Obecne potwierdzone czynniki chorobotwórcze AD obejmują tworzenie się blaszek starczych wywołanych przez nieprawidłowe odkładanie amyloidu-β (aß)i splątania neurofibrilarne lub dystroficzne zapalenie nerwu wywołane przez akumulację tau (Querfurth and LaFerla, 2010; Shinohara et al., 2014). Ponadto na AD mogą mieć również wpływ czynniki genetyczne. Jednak wywołana patogeneza jest nadal niejasna. Do najbardziej rozpowszechnionych leków stosowanych w leczeniu AD należą wzmacniacze neuroprzekaźników, środki przeciw amyloidowe, peptydy neuroprotekcyjne i inne leki (Cacabelos, 2018). W szczególności kilka badań wykazało, że KLF4 odegrał znaczącą rolę w patogenezie AD. W tym przeglądzie skupiamy się na regulacyjnej roli KLF4 w neuroinflammacji, apoptozie neuronalnej, regeneracji aksonalnej i gromadzeniu żelaza, aby wyjaśnić związek między KLF4 a patogenezą AD, co może dostarczyć wglądu w komórkowe i molekularne mechanizmy zaburzeń neurodegeneracyjnych.
charakterystyka biologiczna KLF4
KLF4 jest białkiem jądrowym zawierającym palec cynkowy, wyizolowanym z biblioteki NIH 3T3 i znajdującym się w jądrze komórkowym. Został po raz pierwszy zidentyfikowany i scharakteryzowany przez Shields et al. (1996). Masa cząsteczkowa ludzkiego KLF4 wynosi 55kD i znajduje się na chromosomie 9q31. KLF4 obejmuje 6,3 kb segmentu genu i ma pięć eksonów. Jego region kodujący cDNA koduje polipeptyd składający się z 470 reszt aminokwasowych (jeszcze i wsp., 1998; Ghaleb i Yang, 2017). Karboksyczny koniec klf4 ma region struktury wiążącej DNA zawierający trzy struktury palców cynkowych typu Cys2His2 (C2H2), które są utworzone przez 81 wysoce konserwowanych aminokwasów. Reguluje transkrypcję poprzez wysokie powinowactwo z elementami CACCC i sekwencjami DNA genu docelowego bogatego w GC (Shields and Yang, 1998; Pearson et al., 2008). Większość miejsc wiążących DNA klf4 znajduje się w obszarze palca cynkowego, w tym N-końcowa domena aktywacji transkrypcji dla oddziałujących białek, C-końcowa struktura palca cynkowego dla wiązania DNA i strefa hamowania transkrypcji (Bieker, 2001). KLF4 bierze udział w regulacji ekspresji wielu endogennych genów (Shields i Yang, 1998). Istnieje wysoce zmienna domena regulacyjna transkrypcji na końcu aminowym KLF4. Reszty aminokwasowe znajdujące się między 91 A 117 aminokwasem stanowią domenę aktywacji transkrypcyjnej, która jest bogata w prolinę i serynę, podczas gdy istnieje również domena represji transkrypcyjnej. Dlatego KLF4 ma dwa niekorzystne skutki: aktywację i hamowanie transkrypcji genów (jeszcze et al., 1998; Wei et al., 2006).
podczas rozwoju embrionalnego KLF4 był wyższy w późnym stadium rozwoju embrionalnego. Podczas gdy w dojrzałych tkankach i narządach KLF4 jest wyrażany głównie w przewodzie pokarmowym, jamie ustnej, naskórku skóry, śródbłonku naczyniowym i nerkach, i jest mniej wyrażany w mózgu (Segre et al., 1999; Ghaleb et al., 2011; Liu et al., 2013; Chen et al., 2015; He et al., 2015; Bin et al., 2016). Uważa się, że odgrywa znaczącą rolę w regulacji proliferacji i różnicowania komórek. Poza tym KLF4 może również regulować cykl komórkowy. KLF4 może aktywować P21 w sposób zależny od P53 (Zhang et al., 2000). Ponadto stwierdzono, że komórki KLF4 (–/–) weszły w fazę starzenia wcześniej niż komórki KLF4 ( + / + ), co można wyjaśnić mniejszą ekspresją genu antyoksydacyjnego i wyższym poziomem reaktywnych form tlenu (ROS) w komórkach KLF4 ( – / – ). ROS może zwiększyć ekspresję p53 i P21, a następnie promować uszkodzenie DNA (Liu C. et al., 2015). Stwierdzono, że PRMT5 może podnieść ekspresję KLF4 w poziomie białka. Prmt5 zwiększał transkrypcję p21 i zmniejszał ekspresję bax poprzez hamowanie ubikwitylacji KLF4 (Hu et al., 2015). Ponadto liczne badania wykazały, że KLF4 bierze udział w regulacji apoptozy neuronów(Kong et al., 2016; Cui et al., 2017; Song et al., 2018). Fizjologiczna rola regulacyjna KLF4, o której wiemy, jest wciąż niewielka i potrzebne są dalsze badania.
rola KLF4 w AD
powszechnie wiadomo, że AD charakteryzuje się głównie upośledzeniem pamięci i funkcji poznawczych oraz dysfunkcją wykonawczą (Goedert and Spillantini, 2006). Wiele badań wykazało, że apoptoza neuronalna i dysfunkcja synaptyczna są patologiczną podstawą spadku funkcji poznawczych (Caccamo et al., 2017; Guo et al., 2017; Yoon et al., 2018). Nagromadzone uszkodzenia odkładania się Aß, stresu oksydacyjnego i gromadzenia żelaza mogą prowadzić do dysfunkcji neuronów i apoptozy pacjentów z AD. Kilka badań wykazało, że rola regulacyjna KLF4 wydaje się mieć kluczowe znaczenie dla OUN. Biorąc pod uwagę, że zgłoszono, że KLF4 reguluje apoptozę neuronalną, regenerację synaptyczną, stres oksydacyjny i neuroinflamację, związek między KLF4 a patogenezą AD może być potencjalnym nowym celem leczenia AD.
rola KLF4 w zapaleniu Neuroinflamacyjnym
ilości badań klinicznych wykazały, że Aß może się agregować i jest głównym składnikiem złogów pozakomórkowych tkanki mózgowej pacjentów z AD, co może upośledzać otaczające synapsy i neurony i prowadzić do śmierci neuronalnej. Nieprawidłowe wydzielanie lub nadmierna produkcja Aß prowadzi do patologicznych zmian ad, więc osadzanie aß jest głównym ogniwem AD (Rajmohan i Reddy, 2017). Ponadto badania wykazały, że nadmierne odkładanie się Aß może stymulować komórki glejowe do wydzielania ROS i innych czynników wpływających, co prowadzi do stresu oksydacyjnego. Wiadomo było, że stres oksydacyjny może stymulować produkcję Aß. Dlatego aß i stres oksydacyjny mogą oddziaływać ze sobą i wpływać na postęp AD (Cheignon et al., 2018).
KLF4 został zgłoszony jako potencjalny modulator i ma wielki wpływ na stan zapalny poprzez mediację makrofagów i komórek śródbłonka (ryc., 2014; Kapoor et al., 2015; Yang et al., 2018). W OUN nadmierne i przewlekłe reakcje zapalne mogą powodować uszkodzenie neuronów i neurogliocytów. Niedawno wykazano, że ekspresja KLF4 jest dodatnio skorelowana z wywołanym przez Aß42 neuroinflamacją. W mikroglialnych komórkach BV2 oligomeryczny Aß42 może zwiększać ekspresję KLF4, w której pośredniczy aktywowany P53 (Li L. i wsp., 2017). W stanach zapalnych, takich jak akumulacja Aß, uwalnianie cytokin prozapalnych może być stymulowane w generowaniu AD (Griffin and Barger, 2010). Działanie neurotoksyczności i działanie prozapalne rozpuszczalnych oligomerów Aß42 jest stosunkowo wyższe niż nierozpuszczalne złoża włókien (Selkoe, 1991; Weinberg i in., 2018). Milczenie klf4 jest w stanie przywrócić zapalne zapalenie neuroinflamacyjne za pośrednictwem Aß42, a nadekspresja KLF4 może zaostrzyć zapalne neuroinflamacyjne za pośrednictwem Aß42 (Li L. et al., 2017). Akumulacja Aß indukuje aktywację astrocytów i mikrogleju (Rodríguez et al., 2016). Aktywowane astrocyty mogą nasilać neuroinflammację poprzez uwalnianie czynników prozapalnych, takich jak IL-1, IL-6 i TNF-α (Rubio-Perez and Morillas-Ruiz, 2012; Doméné et al., 2016). Błędne koło reakcji zapalnych ostatecznie prowadzi do dysfunkcji i apoptozy neuronalnej.
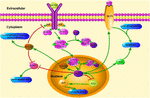
RYSUNEK 1. Schematyczna ilustracja szlaków sygnałowych związanych z KLF4. Liczba ta podkreśla rolę KLF4 w neuroprotekcji i regeneracji aksonów. Strzałki na rysunku wskazują aktywację lub promocję, a linie proste wskazują na powiązane zahamowanie. KLF4, kruppel-like factor 4; STAT3, przetwornik sygnału i aktywator transkrypcji 3; JAK, kinaza Janusa; SOCS3, supresor sygnalizacji cytokin 3; HCP1, białko nośnikowe hemu 1; ERK5, kinaza białkowa aktywowana mitogenem 5.
KLF4 odgrywa kluczową rolę w regulacji sygnałów prozapalnych. W komórkach glejowych indukowana gemfibrozylem aktywacja KLF4 zwiększa supresor sygnału cytokin 3 (SOCS3) poprzez szlak kinazy PI3-AKT (Ghosh and Pahan, 2012). Pośredniczony przez SiRNA knockdown klf4 może złagodzić poziom SOC w astroglii i mikrogleju myszy, co może następnie wpływać na ekspresję genu zapalnego (Kaushik et al., 2010; Ghosh and Pahan, 2012). Ponadto delecja SOCS może promować przetrwanie uszkodzonych neuronów i promować regenerację aksonu (Smith et al., 2009; Sun et al., 2011). A KLF4 pozytywnie reguluje produkcję IL-1β lub innych markerów prozapalnych. Pozytywnie reguluje cyklooksygenazę-2 (Cox-2)i negatywnie reguluje indukowalną syntazę tlenku azotu (iNOS) (Kaushik i wsp., 2013). Ponadto KLF4 jest ważnym czynnikiem regulującym różnicowanie monocytów i potencjalnym celem regulacji immunologicznej (Alder et al., 2008). Dlatego KLF4 może promować neuroinflammation poprzez regulację tych negatywnych regulatorów.
warto wspomnieć, że w modelu choroby Parkinsona KLF4 może promować stres oksydacyjny wywołany MPP+i neurotoksyczność, a następnie zwiększać apoptozę neuronalną i opóźniać proliferację komórek (Chen i wsp ., 2013). Stres oksydacyjny jest nierównowagą między peroksydacją i antyoksydacją. Wolne rodniki mogą powodować zmiany w różnych makrocząsteczkach, co prowadzi do uszkodzenia komórek, starzenia się komórek i uszkodzenia tkanek (Parajuli et al., 2013; Nie et al., 2015). Stres oksydacyjny może nasilać wczesne zapalenie i produkcję Aß, a następnie nasilać AD (Cai et al., 2011). Dlatego KLF4 może być zaangażowany w stres oksydacyjny w AD.
te odkrycia implikują KLF4 kluczową rolę w mediacji neuroinflammation poprzez aktywację mikrogleju i w konsekwencji uwalnianie cytokin prozapalnych. Ma potencjał do zwiększenia neuroinflammation. Do tej pory wiele badań nad patogenezą AD koncentrowało się na neuroinflammacji. Jako potencjalny cel regulacji immunologicznej, KLF4 może promować reakcje zapalne mikrogleju poprzez wpływanie na powiązane negatywne regulatory, co ma wielki wpływ na rozwój AD.
rola KLF4 w apoptozie
zmiany neurodegeneracyjne obejmują stopniową utratę neuronów i synaps w reprezentatywnych regionach mózgu, takich jak kora mózgowa, hipokamp i inne podkorowe regiony. Zaburzenia czynnościowe ośrodkowego układu nerwowego wywołane utratą neuronów są trwałe (Citron, 2010). Trwały stres oksydacyjny może prowadzić do apoptozy neuronalnej (Wu et al., 2010). Wiele badań potwierdziło, że AD jest ściśle związany ze stresem oksydacyjnym (Lee et al., 2012; Yui et al., 2015). Stwierdzono, że przewlekły stres oksydacyjny może nasilać ekspresję fosfolipazy A2 grupy 3 (Pla2g3) w astrocytach i zaburzać równowagę Aß, a w konsekwencji prowadzić do rozwoju AD (Yui et al., 2015).
wiele badań wykazało, że KLF4 odgrywa ważną rolę w hamowaniu rozwoju stresu oksydacyjnego (Shi et al., 2014; Liu C. et al., 2015). Stwierdzono, że KLF4 może promować apoptozę komórek indukowaną przez H2O2, działanie to może być spowodowane zwiększoną ekspresją bax i zmniejszoną ekspresją bcl-2 (Li i wsp ., 2010). Kwercetyna może zmniejszyć ekspresję KLF4 w ludzkich komórkach neuroblastoma SH-SY5Y i zwiększyć ekspresję stosunku Bcl-2/bax. Ponadto kwercetyna może umiarkować szybkość apoptozy komórki SH-5ysy i zmniejszać aktywność enzymu kaspazy-3 (Xi i wsp., 2012). W niedawnym badaniu badano neuroprotekcyjne działanie kinazy białka aktywowanego mitogenem (MAP) 5 (ERK5) przeciwko stresowi oksydacyjnemu. Aktywacja ERK5 może częściowo zmniejszyć śmierć neuronów hipokampa wywołaną H2O2 i zwiększyć neuroprotekcję wywołaną NGF i PC(Su et al., 2014). Nils et al. użył mutanta MEK5 (MEK5D) do badania aktywowanej erk5 transkrypcji i odpowiedzi funkcjonalnych w ludzkich komórkach śródbłonka i zidentyfikował KLF4 jako nowy cel w dalszym ciągu ERK5 (Ohnesorge et al., 2010). Stwierdzono, że nadekspresja KLF4 może hamować odpowiedzi zapalne za pośrednictwem TNF i zmniejszać adhezję leukocytów i apoptozę komórek podstawnych. Wyniki te potwierdzają, że KLF4 ma właściwości przeciwzapalne i anty-apoptotyczne (Ohnesorge et al., 2010). Kolejne eksperymenty wykazały, że zanik mózgowej malformacji jamistej 1 (CCM1) w komórkach śródbłonka aktywuje ERK5 poprzez szlak sygnałowy MEKK3-MEK5 i zwiększa ekspresję KLF4 (Cuttano et al., 2016). ERK5 odgrywa rolę pośrednią w kondycjonowaniu (PC) i czynnik wzrostu nerwów (NGF) reguluje ekspresję KLF4 (Su et al., 2014). Ponadto, wywołane przez RNAi knock-down klf4 może również zmniejszyć neuroprotekcję wywołaną przez NGF lub PC. Nadekspresja KLF4 prowadzi do wyższego stosunku bcl-2 / bax w komórkach obciążonych H2O2 (Su et al., 2014). Nadekspresja KLF4 przyspiesza zmiany w bcl-2 i bax poprzez połączenie z odpowiednim promotorem (Li et al., 2010). Kaskada ERK5 / KLF4 może działać jako ruch obrotowy w różnych szlakach, które chronią neurony przed śmiercią wywołaną stresem oksydacyjnym (Su et al., 2014).
stres oksydacyjny jest uważany za ściśle związany z wieloma chorobami zwyrodnieniowymi. KLF4 odgrywa znaczącą rolę w utrzymaniu stabilności genomowej w stresie oksydacyjnym. KLF4 i ERK5 działają razem, aby chronić neurony przed apoptozą wywołaną stresem oksydacyjnym. Dlatego KLF4 może działać jako cel terapeutyczny, aby działać przeciwko stresowi oksydacyjnemu po aktywacji. Doniesiono, że leki statyny mogą aktywować ERK5, co prowadzi do ekspresji KLF4 i jego zależnych genów (Ohnesorge et al., 2010), ale mechanizm pozostaje niejasny, a geny docelowe związane z klf4 są mniej badane w stresie oksydacyjnym, istnieje potrzeba dalszych badań.
rola KLF4 w regeneracji aksonu
wczesna utrata aksonu jest wspólną cechą chorób neurodegeneracyjnych. Utrata synaptyczna i zaburzenia transportu w AD może powodować zaburzenia poznawcze (Holtzman et al., 2011; Coleman, 2013). Stopień uszkodzenia pamięci deklaratywnej jest związany z gęstością synaptyczną w hipokampie i korze mózgowej. Rozpuszczalne oligomery Aß zmniejszają wychwyt glutaminianu i promują dysfunkcję synaptyczną, zaburzając plastyczność synaptyczną (Li i wsp., 2009). Dlatego szczególnie ważne jest zbadanie, jak naprawić aksony w OUN. W komórkach zwojowych siatkówki aksony mają silną zdolność do wzrostu i regeneracji podczas wczesnego rozwoju, ale w OUN dorosłych ssaków aksony tracą zdolność regeneracji, a neurony mogą umrzeć lub zanik (Goldberg and Barres, 2000; Goldberg et al., 2002).
KLF4 odgrywa ważną rolę w hamowaniu wzrostu aksonów. W embrionalnych RGCs nadekspresja KLF4 może zmniejszyć procent wydłużenia neurytu, długość aksonów i dendrytów oraz rozgałęzienia neurytu. Poza tym stwierdzono, że nadekspresja KLF4 może zmniejszyć długoterminowe postnatalne stopy wzrostu aksonu, ale nie udało się zmniejszyć krótkoterminowych stóp wzrostu aksonu (Moore et al., 2009; Steketee et al., 2014). Późniejsze badania wykazały, że wiązki aksonów myszy KLF4–CKO były grubsze niż myszy kontrolne (Fang et al., 2016). Ponadto usunięcie ekspresji KLF4 podczas rozwoju może zwiększyć potencjał rozrodczy dorosłych RGCs. Ponadto klf4 pozbawiony domeny wiążącej C-końcowy DNA nie miał wpływu na wzrost aksonu. Nie było wpływu na przeżywalność komórek po urazie komórek zwojowych siatkówki, jeśli klf4 był knocking-out (Moore et al., 2009).
KLF4 może również wpływać na regenerację aksonów. Niedawne badanie wykazało, że zmniejszenie ekspresji KLF4 w dorosłych komórkach zwojowych siatkówki promowało regenerację aksonu poprzez szlak jak-STAT3 (Qin i wsp ., 2013). KLF4 zwiększył fosforylację STAT3 i regulował wzrost aksonu za pomocą sygnalizacji JAK-STAT (Qin i Zhang, 2012). Podczas leczenia cytokinami, członkowie rodziny STAT białek są fosforylowane w karboksy-końcowych miejscach tyrozyny i seryny w komórce, tworząc stabilny dimer. Ta modyfikacja zwiększa transkrypcję genów związanych z komórkami (Yuan et al., 2005). Interakcja pomiędzy KLF4 i STAT3 na fosforylację tyrozyny705 indukowaną cytokinami hamuje ekspresję STAT3 poprzez hamowanie wiązania STAT3 z DNA (Qin i in., 2013). Klf4 knockdown oczywiście poprawia regenerację axon w komórkach zwojowych siatkówki po uszkodzeniu nerwu wzrokowego i zapobiega uszkodzeniu nerwu po łagodnym uszkodzeniu mózgu. W działaniach tych pośredniczy zmniejszenie stężenia p-p53 i zwiększenie stężenia pstat3. KLF4 pozytywnie reguluje apoptozę neuronalną poprzez szlaki p53 i jak-STAT3, a klf4 negatywnie reguluje naprawę aksonalną poprzez szlak JAK-STAT3 (Cui et al., 2017).
dlatego postawiliśmy hipotezę, że w AD regenerację aksonalną można osiągnąć poprzez zmianę ekspresji KLF4 lub zmianę wewnątrzkomórkowych szlaków sygnałowych i kontrolowanie progresji AD poprzez zmniejszenie brakujących aksonów lub zmniejszenie dysfunkcji aksonów. Jednak jak wykorzystać czynnik transkrypcyjny KLF4 w potencjalnych terapeutach nadal wymaga dalszych poszukiwań.
rola KLF4 w akumulacji żelaza
żelazo jest szeroko spotykane w systemach biologicznych, metaloproteinazy związane z żelazem odgrywają kluczową rolę w transporcie tlenu, przenoszeniu elektronów i katalizowaniu reakcji biochemicznych (Aisen et al., 2001). Jednak każdy nadmiar żelaza poza normalnym zakresem fizjologicznym może zaszkodzić zdrowiu ludzkiemu (Adlard and Bush, 2006). Badania wykazały, że zawartość żelaza w hipokampie jest negatywnie skorelowana z wynikami testów pamięci (Ding et al., 2009). Zwiększone obciążenie żelazem w mózgu przyspiesza powstawanie blaszek Aß i hiperfosforylowanych splątań tau, jednocześnie zwiększając stres oksydacyjny(Peters et al., 2015). Żelazo, które ma wysoki stopień przepuszczalności, wspomaga wzrost nerwów i połączenia między komórkami podczas rozwoju mózgu (Dallman i Spirito, 1977).
ostatnie badania wykazały, że stres fizjologiczny spowodował aktywację szlaku sygnałowego KLF4-HCP1 i zwiększony wychwyt hemu (Li H. i wsp., 2017). Hem stanowi 95% funkcjonalnego żelaza w organizmie człowieka. Jest jednym z głównych składników oksygenazy hemu (Hooda et al., 2014; Kurucz et al., 2018). Zwiększenie aktywności oksygenazy – 1 może opóźnić utlenianie starzejącego się mózgu (Verdile et al., 2015; Serini and Calviello, 2016; Kurucz et al., 2018). Ma to efekt ulgi w reklamie. Stres fizjologiczny powoduje wzrost poziomu glikokortykosteroidów, glikokortykosteroidy zwiększają ekspresję białka nośnika hemu 1 (HCP1) poprzez KLF4, a następnie HCP1 Promuje wychwyt hemu (Li H. i wsp., 2017). Glikokortykosteroidy i KLF4 regulują razem geny przeciwzapalne, a komórki o niskiej zawartości glikokortykosteroidów nie mogą w pełni indukować ekspresji KLF4(Sevilla i wsp ., 2015). Indukowany przez KLF4 wzrost spożycia hemu prowadzi do gromadzenia się żelaza w mózgu. Żelazo sprzyja uwalnianiu ROS (Tronel et al., 2013). Pierwiastek żelaza zwiększa stres oksydacyjny mózgu u szczurów pod wpływem stresu psychicznego (Yu et al., 2011). Dlatego HCP1 może być regulowany przez klf4 i glikokortykosteroidy razem. Zwiększenie HCP1 zwiększa wychwyt hemu, co prowadzi bezpośrednio do gromadzenia się żelaza w mózgu, nasila utlenianie, zwiększa apoptozę lub dysfunkcję i pogarsza uszkodzenie mózgu.
ogólnie przyjmuje się, że upośledzenie pamięci i uczenia się są głównymi objawami AD. Duża liczba danych klinicznych wykazała, że obciążenie płytki nazębnej Aß i reakcja akumulacji żelaza na rozwój zaburzeń uczenia się i poznawczych w AD (van Bergen et al., 2018). Ostatnio opublikowane dane sugerują, że Wysokie dawki żelaza zwiększają odkładanie się Aß i osłabiają uczenie się i pamięć u myszy (Guo et al., 2013). Badania kliniczne wykazały, że mikroglej zawierający żelazo znajduje się w hipokampie pacjentów z AD pod rezonansem magnetycznym (Zeineh et al., 2015). Mikroglej pozyskuje żelazo z przenoszących lub nie przenoszących, zewnątrzkomórkowych i wewnątrzkomórkowych źródeł (McCarthy et al., 2018). Selektywna i trwała ekspresja KLF4 może być indukowana w jądrze i cytoplazmie niedokrwiennego hipokampa reaktywnych astrocytów (Park et al., 2014). Badania wykazały, że KLF4 działa jako represor transkrypcyjny. Reguluje ekspresję ELK-3, a następnie ELK-3 hamuje ekspresję HO-1 (Tsoyi et al., 2015). Oksygenaza hemu-1 (HO-1) jest białkiem stresowym, które rozkłada hemu do bilirubiny, wolnego żelaza i tlenku węgla. Regulacja HO-1 w astrocytach może prowadzić do nieprawidłowego odkładania żelaza i dysfunkcji mitochondriów w mózgu, co prowadzi do zmniejszenia zdolności poznawczych (Schipper, 1999, 2004). Dlatego KLF4 może być zaangażowany w proces akumulacji żelaza w astrocytach, nasilając utlenianie w AD i pogarszając uszkodzenie mózgu.
wniosek
klf4 jest powszechnie znany jako odgrywający kluczową rolę w regulacji proliferacji komórek, apoptozy i różnicowania. Wcześniejsze badania koncentrowały się na regulacji KLF4 w kilku ważnych procesach neurofizjologicznych, w tym neuroinflammacji, neuroprotekcji i regeneracji synaptycznej. Ostatnio stwierdzono, że KLF4 odgrywa ważną rolę w patogenezie AD. W tym artykule dokonujemy przeglądu roli KLF4 w neuroprotekcji i neurogenezie w AD.
KLF4 jest nie tylko regulatorem regulacji proliferacji i różnicowania komórek, ale także potencjalnym celem regulacji odpowiedzi immunologicznych. KLF4 może regulować negatywne czynniki zapalne i promować odpowiedź zapalną i mieć wielki wpływ na ekspresję astrocytów jądrowego mikrogleju. Ponadto KLF4 i ERK5 mogą działać razem, aby wywierać działania neuroprotekcyjne. Ponadto regenerację aksonu można osiągnąć poprzez zmianę zawartości określonych czynników transkrypcyjnych, wewnątrzkomórkowych inhibitorów lub zmianę wewnątrzkomórkowych szlaków sygnałowych. Wyeliminowanie KLF4 może zwiększyć regenerację axon i przyspieszyć tempo wzrostu axon. Redukcja ekspresji KLF4 promuje regenerację aksonu poprzez szlak jak-STAT3, a KLF4 Promuje szlak jak-STAT3 do dalszej regeneracji aksonu. Dlatego klf4 może być zaangażowany w proces przeciwzapalny, przeciw apoptozie, regeneracji aksonu i gromadzenia żelaza w OUN, co odgrywa kluczową rolę w generowaniu AD. Wyniki te sugerują, że KLF4 stanowi potencjalny cel terapeutyczny dla AD. Jednak głębokie komórkowe i molekularne mechanizmy wpływu KLF4 na AD pozostają niejasne i potrzebne są dalsze badania.
autor
rękopis napisał ZQC, XHZ i YJ. SHG i JYL zmodyfikowali ramy rękopisu. BJL i RJC dostarczyły krytyczne poprawki. Wszyscy autorzy zaakceptowali ostateczną wersję manuskryptu do przesłania.
finansowanie
ta praca była wspierana przez granty z Natural Science Foundation Of China (NSFC) (81871070, 31571126, 31471120) i Jilin Science and Technology Agency (Grant nr 20180519003jh, 20180414051gh, 20170414034gh i 20180414050gh).
Oświadczenie o konflikcie interesów
autorzy oświadczają, że badania zostały przeprowadzone przy braku jakichkolwiek relacji handlowych lub finansowych, które mogłyby być interpretowane jako potencjalny konflikt interesów.
Cacabelos, R. (2018). Czy w ciągu ostatnich 5 lat nastąpiła poprawa w odkryciu leku na chorobę Alzheimera. Exp. Opin. Lek Discov. 13, 523–538. doi: 10.1080/17460441.2018.1457645
PubMed Abstract / CrossRef Pełny tekst / Google Scholar
Liu, Y., Shi, J., and Chen, X. (2015). Identyfikacja nowych celów dla sezonowego alergicznego nieżytu nosa podczas i poza sezonem pyłkowym za pomocą analizy mikromacierzy. Acta Otolaryngol. 135, 1330–1336. doi: 10.3109/00016489.2015.1067906
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Serini, S., and Calviello, G. (2016). Redukcja stresu oksydacyjnego / nitrozacyjnego w mózgu i jego udział w neuroprotekcyjnym działaniu N-3 PUFA w chorobie Alzheimera. Curr. Alzheimer Res. 13, 123-134. doi: 10.2174/1567205012666150921101147
PubMed Abstrakt / CrossRef Pełny tekst / Google Scholar