wprowadzenie
Gen cząsteczki adhezji komórek L1 (L1CAM) jest cząsteczką adhezji komórek neuronalnych należącą do nadrodziny immunoglobulin; posiada kluczowe funkcje w rozwoju układu nerwowego (Itoh and Fushiki, 2015). Mutacje w L1CAM były związane z zespołami neurologicznymi związanymi z X, które są podsumowane jako choroby L1. Są one klasyfikowane w następujący sposób: Wodogłowie X-linked (XLH) z powodu zwężenia akweduktu Sylviusa (HSAS), zespołu masy (niepełnosprawność intelektualna, afazja, tasowanie chodu, przytłumione kciuki), paraparezy spastycznej typu 1 (SP1) i agenezy X-linked corpus callosum (ACC) (Weller and Gartner, 2001; Itoh and Fushiki, 2015).
w HGMD® Professional 2019.2 (https://portal.biobase-international.com/hgmd/pro/all.php) zgłoszono około 282 mutacje powodujące chorobę (DMS) w genie L1CAM. Zmiany w genie L1CAM są zróżnicowane; analizy danych mutacji od 282 pacjentów ujawniają 51% mutacji missense i nonsens, 25% delecji, 5% insercji i 19% zmian miejsca splicingu, ale ciche mutacje w L1CAM o potencjale patogennym były rzadkie, a ciche mutacje były często ignorowane, szczególnie w wykrywaniu sekwencjonowania całego eksomu (WES).
w tym badaniu, przy użyciu WES, przebadaliśmy DNA płodu chińskiej kobiety ciężarnej, która zgłosiła pięć ciągłych ciąż z wodogłowiem płodu; znaleźliśmy tylko nową milczącą mutację c. 453g > T (P. Gly151 = ) w genie L1CAM. Co ciekawe, poprzez dalszą analizę, wykazaliśmy, że cicha mutacja stworzyła potencjalną sekwencję konsensusu miejsca splicingu 5′, co skutkowałoby wewnętrzną delecją 72 bp z eksonu 5 i 24 aminokwasów białka L1CAM.
Prezentacja przypadku
do naszej kliniki skierowano 28-letnią zdrową kobietę po czterech dobrowolnych przerwach ciąży z powodu wodogłowia płodu w innych szpitalach. Wszystkie płody były płci męskiej. Po przybyciu do naszego szpitala (Women ’ s Hospital, School Of Medicine, Zhejiang University, Zhejiang, Chiny) była już w piątej ciąży W 24 tygodniu ciąży, z wodogłowiem płodu na podstawie badań obrazowych. Aby zbadać przyczynę genetyczną, próbki krwi płodu przeprowadzono w 26 tygodniu ciąży. Konwencjonalne badania cytogenetyczne przeprowadzono zarówno dla próbek płodowych, jak i rodzicielskich, a próbkę płodową analizowano dalej za pomocą tablicy polimorfizmu pojedynczego nukleotydu (SNP) i WES.
badanie to zostało przeprowadzone zgodnie z zaleceniami Komitetu etycznego szpitala kobiet, School Of Medicine Zhejiang University, a wszyscy uczestnicy tego badania uzyskali świadomą zgodę zgodnie z deklaracją Helsińską. Protokół badania został zatwierdzony przez Komisję Rewizyjną szpitala kobiet, School Of Medicine, Zhejiang University w Chinach.
materiały i metody
kariotyp i tablica SNP
kariotypy krwi pępowinowej płodu i krwi pępowinowej obwodowej oznaczono konwencjonalnym kariotypowaniem co najmniej 30 limfocytów krwi, które zostały zatrzymane w metafazie przez kolchicyny. G-banding kariotypów hodowanych komórek przeprowadzono na poziomie 320-400-pasm z rozdzielczością około 10 Mb. Tablica SNP została wykonana przez tablicę CytoScan™ HD (Affymetrix, USA) zgodnie z instrukcją producenta, z około 2 600 000 markerami, w tym 750 000 sond SNP i 1 900 000 sond nie polimorfizmu dla kompleksowego pokrycia całego genomu. Dane analizowano za pomocą oprogramowania do analizy chromosomów (Chas) (Affymetrix, Santa Clara, CA) w oparciu o zespół GRCh37/hg19. Próg sprawozdawczy wyniku liczby kopii został ustalony na 500 kb z liczbą znaczników ≥50 dla zysków i na 200 kb z liczbą znaczników ≥50 dla strat.
sekwencjonowanie całego Egzomu
główna część WES została dostarczona przez Beijing Genomics Institute. Genomowy DNA został wyekstrahowany przez zestaw krwi DNeasy (Qiagen, CA), a następnie został fragmentowany przez Covaris LE220 (Massachusetts, USA) w celu wygenerowania sparowanej biblioteki (200-250 bp). Wszystkie amplifikowane biblioteki przeprowadzono na platformie BGISEQ-500, jednoniciowy DNA zmieszano z MGIEasy™ DNA Library Prep Kit V1 (BGI, Shenzhen, Chiny), a następnie zsekwencjonowano przy użyciu chemii 100SR z BGISEQ-500Rs high-through sequencing Kit (BGI, Shenzhen, Chiny).
czyste odczyty (o długości 90 bp) uzyskane z ukierunkowanego sekwencjonowania i filtrowania zostały następnie wyrównane do odniesienia do ludzkiego genomu (hg19) przy użyciu pakietu oprogramowania Wielowizyjnego Burrows-Wheeler Aligner (BWA) (Li i Durbin, 2009). Po wyrównaniu, pliki wyjściowe zostały wykorzystane do wykonania sekwencjonowania pokrycia i analizy głębokości regionu docelowego, warianty pojedynczego nukleotydu (SNVs), i wywołanie indel, użyliśmy oprogramowania GATK do wykrywania SNVs i indels (McKenna et al., 2010), wszystkie SNV i indele były filtrowane i szacowane za pomocą wielu baz danych, w tym National Center for Biotechnology Information (NCBI) Single-nucleotide Polymorphism Database (dbSNP), HapMap, 1000 Genomes Project dataset i bazy danych 100 chińskich zdrowych dorosłych. Użyliśmy Condel, SIFT, PolyPhen-2, LRT, Mutation Taster i Phyllop, aby przewidzieć efekt wariantów. Warianty chorobotwórcze są oceniane zgodnie z protokołem wydanym przez American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015). Human Gene Mutation Database (HGMD) użyto do przesiewania mutacji. Wszystkie potencjalne warianty chorobotwórcze zwalidowano przy użyciu metod sekwencjonowania Sangera.
ekstrakcja RNA, PCR i sekwencjonowanie
jednojądrzaste komórki krwi obwodowej (Pmbcs) i jednojądrzaste komórki krwi pępowinowej (CBMCs) wyizolowano przez rozdzielenie gradientu gęstości Ficolla. Całkowity RNA ekstrahowano z PMBCs i CBMCs przy użyciu Trizolu (Takara, Japonia). Wyekstrahowane całkowite RNA zostały poddane odwrotnej transkrypcji za pomocą zestawu RT (Takara, Japonia). PCR wykonano przy użyciu GoldStar Best Master Mix (CWBIO, Pekin). Sekwencje starterowe są wymienione: L1CAM-DNA-5F, CCCACCCGTCCTTTCCTA; L1CAM-DNA-5R, CGCTCGTCCTGCTTGATGT; L1CAM-mRNA-4-6-F, GGTGTCCACTTCAAACCAA; oraz l1cam-mRNA-4-6-R, GCGGCTTCCTGTCAATCA. Sekwencjonowanie Sangera przeprowadzono analizatorem DNA ABI 3130.
wyniki
do naszej kliniki skierowano 28-letnią zdrową kobietę po czterech dobrowolnych przerwach ciąży z powodu wodogłowia płodu. Wszystkie płody były płci męskiej (rycina 1A). Rodowód rodziny zdawał się wskazywać na XLH. Była już w piątej ciąży w 26 tygodniu ciąży. Ventriculomegaly płodu wykryto przez USG płodu i MRI, które konsekwentnie wykazały obecność wodogłowia. Wykazały one, że obustronna komora mózgowa i trzecia komora były oczywiście rozszerzone, a w śródmózgowej i agenezie ciała modzelowatego wystąpiło ciężkie wodogłowie(ryc. 1B).
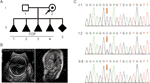
Rys. 1 (A) Rodowód rodziny. Góra, przerwanie ciąży. B) badania obrazowe płodu. USG płodu i rezonans magnetyczny płodu wykazały, że płód miał ciężkie wodogłowie. (C) analiza sekwencji genomowego DNA od członków rodziny. Genotypy L1CAM były typu dzikiego, c.453g > T Het i c. 453g > T Hom, w I:1 (mąż), I: 2 (kobieta w ciąży) I II:5 (płód). Mutacja jest oznaczona czerwonymi strzałkami.
aby zbadać możliwą przyczynę genetyczną, przeprowadziliśmy analizę kariotypu i tablicę SNP w celu analizy próbek krwi płodu i nie znaleźliśmy żadnych pozytywnych wyników. Wynik neowaskularyzacji choroidalnej (CNV) został zdeponowany w Omnibusie ekspresji genu (GEO); numer przystąpienia to GSE133063, jak załączono poniżej (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
następnie wykryliśmy płód przez Wesa. Strategia analityczna dla znalezienia prawdopodobnej identyfikacji wariantu chorobotwórczego została przedstawiona na rysunku S1. Listę wariantów (tabela S1) uzyskano poprzez badanie przesiewowe częstotliwości wariantów, statusu mutacji i trybu dziedziczenia. Biorąc pod uwagę geny związane z wodogłowiem (HP:0000238, http://compbio.charite.de/hpoweb/showterm?id=HP:0000238#id=HP_0000238) (tabela S2), nie stwierdzono dodatkowych istotnych mutacji, z wyjątkiem cichej mutacji c.453g > T w eksonie 5 genu L1CAM (NM_000425.3). c. 453g > T nie został zgłoszony w HGMD i ClinVar i nie został znaleziony w dbSNP, gnomAD i innych zestawach danych. Zgodnie ze standardami i wytycznymi acmg (Richards et al., 2015), nie osiągnął jeszcze kryterium „patogennej” lub „prawdopodobnej patogennej”, ale nie było innych potencjalnych mutacji; nie mieliśmy wyboru, jak tylko dokonać dalszej analizy stwierdzonej mutacji cichej.
zgodnie z tradycyjnym myśleniem, ta podstawienie miało miejsce w trzeciej zasadzie w kodonie 151, który koduje glicynę, tworząc w ten sposób mutację neutralną (P. Gly151=). Ten wariant został potwierdzony w DNA wyekstrahowanym z krwi pępowinowej płodu i krwi obwodowej w parze przez sekwencjonowanie Sangera (Fig. 1C). Kobieta nosiła mutację heterozygotyczną, a jej mąż był genotypem typu dzikiego.
z mutacją (http://www.mutationtaster.org/), c. 453g > t oceniono jako ” wywołującą chorobę.”Wykazano, że funkcje białkowe mogą być naruszone i miejsce łączenia może zostać zmienione; byliśmy ciekawi potencjalnych efektów splicingu funkcji L1CAM w tej cichej mutacji. Cicha mutacja została przetestowana przy użyciu następujących produktów oprogramowania online: NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) i NNSplice (http://www.fruitfly.org/seq_tools/splice.html); przewidywano również utworzenie 5′ potencjalnego miejsca splicingu w L1CAM C.453g > t mutacja przy użyciu produktów oprogramowania (rysunek S2). Wyniki wykazały, że ta cicha mutacja wytworzyła potencjalne miejsce splotu 5 ’ 72 bp przed normalnym miejscem splotu eksonu 6/IntronA 6 (Fig. 2A). Jeśli tak jest, możemy znaleźć zmianę długości l1cam messenger RNA (mRNA) między I:2 I II:5 (Fig.2A). RT-PCR wykonano za pomocą starterów zaprojektowanych do wzmacniania eksonów 4-6 w mRNA L1CAM. Rzeczywiście, wyniki wykazały krótki zakres obcinania w PCR cDNA płodu (II:5), podczas gdy pasmo amplifikowane z l1cam mRNA zawierało oczekiwane długie pasmo w PCR cDNA męża (I:1) i długie / krótkie pasma u kobiety ciężarnej cDNA PCR (I: 2) (rys. 2b). Bezpośrednie sekwencjonowanie amplifikowanego fragmentu wykazało, że delecja dotyczyła ostatnich 72 bp eksonu 5 u męskiego płodu cDNA (kobieta była nosicielem) (Fig.2C). Mamy kluczowe dowody chorobotwórcze.
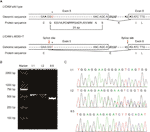
2 (A) schematyczna reprezentacja organizacji eksonu 5, IntronA 6 i eksonu 6 w L1CAM. B) analiza RT-PCR eksonów 5 i 6 cDNA L1CAM z jednojądrzastych komórek krwi obwodowej (pmbcs) i jednojądrzastych komórek krwi pępowinowej (CBMCs). Elektroforeza w żelu agarozowym produktów RT-PCR wytworzonych z I:1 (mąż), I: 2 (kobieta w ciąży) I II:5 (płód). C) analiza sekwencji produktu RT-PCR z PMBCs pary i CBMCs płodu.
ta cicha mutacja dała 24 aminokwasy białka L1CAM (reszty 151-174); Lys (K) został podstawiony przez Glu (E) w kodonie 175 (Fig. Stwierdzono wyrównanie wielu sekwencji białka L1CAM u kilku gatunków i zachowanie brakujących aminokwasów w L1CAM u ssaków: Homo sapiens, Pan troglodytes, Bos taurus, Mus musculus i Rattus norvegicus (rysunek 3A). Mutacje typu dzikiego i c.453g > t splicing białka l1cam były przewidywane przez oprogramowanie Cphmodels-3.2 Server (http://www.cbs.dtu.dk/services/CPHmodels/) (rysunek 3B). Immunoglobulinopodobna (Ig-like) domena 2 (pozostałości 134-230) mutacji dzikiego typu i splicingu białek l1cam pokazano na fig. 3C.l1cam c. 453G > mutacja splicingu t zmieniła strukturę białka, zwłaszcza domenę IG-podobną 2

3 (A) wyrównanie wielu sekwencji białek L1CAM w obrębie gatunków. L1CAM C.453G > t spowodowało brak 24 aminokwasów białka L1CAM (reszt 151-174) w konserwowanym regionie aminokwasowym u różnych gatunków. Czarna kolumna pokazuje brakujące aminokwasy. B) struktury typu dzikiego i c. 453g > t splicing mutacji białka L1CAM zgodnie z przewidywaniami oprogramowania Cphmodels-3.2 Server. C) struktury immunoglobulinopodobnej (IG-like) domeny 2 (reszt 134-230) dzikiego typu i splicingu mutacji białka l1cam.
dyskusja
Ciche mutacje były często wykrywane przez WES, ale niewystarczająca uwaga została zwrócona, co doprowadziło do pominięcia DMs. W tym badaniu zatrudniliśmy Wesa do zbadania genetycznej przyczyny chińskiej rodziny z wodogłowiem, ale znaleźliśmy tylko nową milczącą mutację w L1CAM, co zmusiło nas do dalszej analizy. Na szczęście udowodniliśmy, że silent mutation stworzył nową stronę 5′ splice i był DM.
mutacje w L1CAM mogą powodować X-linked L1 disease, ale objawy kliniczne są zmienne; mutacje wytwarzają nieoczekiwane fenotypy. W badaniu pięć cierpiących płodów to mężczyźni, co jest zgodne ze wzorem dziedziczenia. USG płodu i MRI wykazują typową chorobę L1, W tym XLH i agenezę ciała modzelowatego. Poprawia nasze zrozumienie korelacji genotypowo-fenotypowej L1CAM.
L1CAM c.453G > T (P. Gly151 = ) początkowo uważano, że nie ma wpływu na sekwencję białka. Ale inne nieme mutacje, c.924C > T (P. Gly308 = ) i c.645C > T (P. Gly215 = ), w genie L1CAM zostały zgłoszone jako DMs (Du et al., 1998; Vos et al., 2010). C.Mutacja 924c > t spowodowała aktywację nowego miejsca splotu 69 bp 5′ do normalnego miejsca splotu dawcy eksonu 8/IntronA 8 i została uznana za miejsce „chorobotwórcze”wodogłowia (Du et al., 1998). Dla c. 645C > T w L1CAM usunięto 51 bp wraz z aktywacją nowego połączenia dawcy eksonu 6/IntronA 6 (Vos et al., 2010). Nasze obecne badanie było podobne; mutacja c. 453g > t stworzyła potencjalne miejsce splotu 5′ przed normalnym miejscem splotu eksonu 5/IntronA 5. Wszystkie te ciche mutacje stworzyły nowe miejsca łączenia dawców, w wyniku czego ekson został pominięty. Przypomniało nam to o zwróceniu szczególnej uwagi na te ciche mutacje, które mogą wpływać na splicing białek.
jako glikoproteina przezbłonowa i członek nadrodziny immunoglobulin cząsteczek adhezyjnych komórek, białko L1CAM może oddziaływać na powierzchni komórki z wieloma różnymi glikoproteinami, a wiązanie homofilne jest prawdopodobnie głównym sposobem interakcji (Wei And Ryu, 2012). Badania nad strukturą krystaliczną domen 1-4 podobnych do Ig u neurofascyny sugerowały, że wiele patologicznych mutacji L1 wpływa na zachowane reszty aminokwasowe w tych domenach i koliduje z interakcjami homofilnymi (Liu et al., 2011), zwłaszcza jak zweryfikowane przez badania funkcji Ig-like domeny 2 (Zhao et al., 1998). W naszych badaniach spekulowaliśmy, że l1cam c.453g > t zmienia domenę podobną do Ig w zewnątrzkomórkowej części białka l1cam, prowadząc do nieprawidłowej interakcji zewnątrzkomórkowej, nie rozpoczynając inicjacji poniżej szlaku sygnałowego. Dalsze badania poświęcone spektrometrii masowej tego wariantu L1CAM wyjaśniłyby dokładnie, jaki zespół molekularny jest wytwarzany w komórce.
podsumowując, za pośrednictwem WES, zgłosiliśmy nową milczącą mutację c. 453g > T w L1CAM, która wytwarza 5′ miejsce splotu odpowiedzialne za wodogłowie. Przewidywano, że ten nieprawidłowy wariant białka zmieni domenę 2 podobną do Ig, co może wpływać na Wiązanie homofilne białka L1CAM. Ponadto przeprowadziliśmy prenatalną diagnostykę genetyczną kobiety ciężarnej, która zgłosiła pięć ciągłych ciąż z wodogłowiem. W międzyczasie zasugerował, że nie należy ignorować niektórych milczących mutacji wykrytych w WES; konieczne było przepowiadanie tych mutacji. Dostarczyła ona nowych genetycznych podstaw do diagnostyki prenatalnej i przedimplantacyjnej diagnostyki prenatalnej wodogłowia.
dostępność danych
publicznie dostępne zbiory danych zostały przeanalizowane w tym badaniu. Dane te można znaleźć tutaj: GSE133063 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133063).
Oświadczenie o etyce
badania z udziałem uczestników ludzkich zostały zweryfikowane i zatwierdzone przez Komisję Rewizyjną szpitala kobiet, Szkoły Medycznej, Uniwersytetu Zhejiang W Chinach. Pisemną świadomą zgodę na udział w badaniu udzielił opiekun prawny/najbliższy krewny uczestników. Pisemna, świadoma zgoda została uzyskana od osoby (osób) i opiekuna prawnego (osób) nieletnich/najbliższych krewnych na publikację wszelkich potencjalnie możliwych do zidentyfikowania obrazów lub danych zawartych w tym artykule.
autorzy
Ys, yli, MC, YLu, YQ i YY przeprowadzili eksperymenty. Ys przygotował dane. MC i YLi przeanalizowali dane WES. YLu i YQ przeprowadziły analizę kariotypu i SNP array. YY zwerbował próbki. Hl i FL zapewniły badania obrazowe. Ys I MD napisali manuskrypt. Wszyscy autorzy przeczytali i zatwierdzili ostateczny manuskrypt.
finansowanie
badanie to było wspierane przez National Natural Science Foundation Of China (Grant nr 81801441), kluczowy program badawczo-rozwojowy prowincji Zhejiang (Grant nr 81801441). 2019c03025), Narodowy kluczowy program badawczo-rozwojowy Chin (Grant nr 2016yfc1000703) oraz medyczna Fundacja Badań Naukowych prowincji Zhejiang (Grant nr 2014kya246).
Oświadczenie o konflikcie interesów
autorzy oświadczają, że badanie zostało przeprowadzone przy braku jakichkolwiek relacji handlowych lub finansowych, które mogłyby być interpretowane jako potencjalny konflikt interesów.
podziękowania
dziękujemy pacjentom włączonym do tego badania. Dziękujemy Dr. Jiong Gao (BGI Genomics, BGI-Shenzhen, Shenzhen 518083, Chiny) za pomoc w przygotowaniu tego rękopisu.
Materiały uzupełniające
Materiały uzupełniające do tego artykułu można znaleźć w Internecie pod adresem: https://www.frontiersin.org/articles/10.3389/fgene.2019.00817/full#supplementary-material
rysunek S1 / Strategia analityczna dla znalezienia prawdopodobnej identyfikacji wariantu chorobotwórczego przez WES.
rysunek S2 / miejsca łączenia dawców przewidywane przez NetGene2 i NNSplice.
Table S1 / List of variants through the screening of variants frequencies, mutation status, and inheritance mode.
tabela S2 / lista genów związanych z wodogłowiem (eksport do HP: 0000238).
Du, Y. Z., Dickerson, C., Aylsworth, A. S., Schwartz, C. E. (1998). Cicha mutacja, C924T (G308G), w genie L1CAM powoduje X linked hydrocephalus (HSA). J. Med. Genet. 35 (6), 456–462. doi: 10.1136 / jmg.35.6.456
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
Itoh, K., Fushiki, S. (2015). Rola L1cam w kortykogenezie myszy i patogenezie wodogłowia. Pathol. Int. 65 (2), 58–66. doi: 10.1111 / pin.12245
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
Li, H., Durbin, R. (2009). Szybkie i dokładne dopasowanie do krótkiego odczytu z transformatą Burrowsa-Wheelera. Bioinformatyka 25 (14), 1754-1760. doi: 10.1093/bioinformatyka / btp324
PubMed Abstract / CrossRef Full Text / Google Scholar
Liu, H., Focia, P. J., He, X. (2011). Homofilny mechanizm adhezji neurofascyny, członka rodziny cząsteczek adhezyjnych komórek nerwowych L1. J. Biol. Chem. 286 (1), 797–805. doi: 10.1074 / jbc.M110.180281
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297-1303. doi: 10.1101 / gr.107524.110
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi: 10.1038 / gim.2015.30
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Vos, Y. J., de Walle, H. E. K., Bos, K. K., Stegeman, J. A., Berge, A. M., Bruining, M., et al. (2010). Genotyp-fenotyp korelacje w L1 syndrome: a guide for genetyc counselling and mutation analysis. J. Med. Genet. 47 (3), 169–175. doi: 10.1136 / jmg.2009.071688
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
(2012) Homofilne oddziaływanie cząsteczek adhezyjnych z rodziny L1. Exp. Mol. Med. 44 (7), 413–423. doi: 10.3858 / emm.2012.44.7.050
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
Weller, S., Gartner, J. (2001). Genetyczne i kliniczne aspekty wodogłowia związanego z X( choroba L1): mutacje w genie L1CAM. Hum. Mutat. 18 (1), 1–12. doi: 10.1002 / humu.1144
PubMed Abstract | CrossRef Full Text | Google Scholar
Zhao, X., Yip, P. M., Siu, C. H. (1998). Identification of a homophilic binding site in immunoglobulin-like domain 2 of the cell adhesion molecule L1. J. Neurochem. 71 (3), 960–971 doi: 10.1046/j.1471-4159.1998.71030960.x.
PubMed Abstract | CrossRef Full Text | Google Scholar