tekniken för DNA-sekvensering utvecklades redan 1977 tack vare Frederick Sanger. Det tog lite längre tid innan det var möjligt att sekvensera ett fullständigt genom. Detta beror på att vi behövde en lämplig matematisk modell och massiv beräkningskraft för att montera miljoner eller miljarder små läsningar till ett större komplett genom. Dagens beräkningskraft och programvara är den största skillnaden mellan vad som brukade ta år av arbete i början av 2000-talet och vad som bara tar några timmar idag. Algoritmen du valde att göra detta är” holy grail ” av monteringstekniken. Dessa algoritmer innehåller en av de mest kända variablerna som är kända i matematiska modeller, k-meren.
ursprunget till k-meren och den matematiska modellen som omger den kommer från en schweizisk matematiker från 1735 Leonhard Euler, som är känd som fadern till den matematiska funktionen. En holländsk matematiker Nicolaas de Bruijn anpassade Eulers ideer för att hitta en cyklisk sekvens av bokstäver tagna från ett givet alfabet för vilket alla möjliga ord av en viss längd visas som en rad på varandra följande tecken i den cykliska sekvensen exakt en gång.
de Bruijns algoritm anpassades av molekylärbiologer, som många år senare mötte ett motsvarande problem: hur man monterar DNA-sekvenser. Således använder forskare över hela världen nu de Bruijn-grafen och variabeln k.
tillämpning av k-mers för att montera DNA-sekvenser
i några få ord innebär de novo genommontering att man förbinder på varandra följande små DNA-läsningar och slutar med större sekvenser. För att generera en de Bruijn-Graf (se figuren nedan) måste nukleotiderna vid kanten av varje läsning överlappa kanten på en andra (och så vidare). Det slutliga målet är att skapa ett på varandra följande toppunkt, vilket (potentiellt) kommer att resultera i stora DNA-fragment.
du måste fragmentera dina läsningar i k-mers, som är ett specifikt antal nukleotider som överlappar varandra. Med k-mer kan du skapa en unik sekvens från många små. Varje unik k-Mer-sekvens identifieras och extra kopior elimineras. Denna aspekt av k-mers låter dig övervinna en av nackdelarna med nästa generations sekvensering — att få läsningar som representerar genomiska regioner med olika frekvenser (dvs att få mycket små läsningar från en region). Användningen av k-mers eliminerar sekvenser som upprepas mer än en gång på grund av ojämn sekvenstäckning. Tänk dock på att en låg k-Mer-storlek ökar chanserna för överlappning av nukleotider, medan ett större värde minskar dem.
dagens de novo–monteringsteknik är effektivare när du använder bibliotek med stora läsningar (dvs 1.000-10.000 bps) i kombination med mindre (100-200 bps). Program kan använda K-värdet och k-mers för att montera korta läsningar. Dessa kan sedan införlivas och verifieras av större för att hamna i mer exakta contigs.
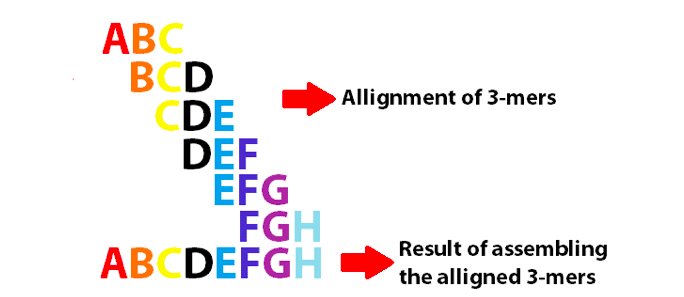
exempel på en de Bruijn-graf med 3-mers för att montera de 8 första bokstäverna i det engelska alfabetet. Observera att dessa 3-mers överlappar varandra som k-1.
ju mer du vet desto mer kan du uppnå i DNA-montering
det finns specifika tips du behöver tänka på innan du använder de Bruijn-grafer i din monteringsmetod och väljer den mest lämpliga k-Mer-storleken. Genom att utnyttja dessa kan du skapa bättre resultat.
- först och främst, och kanske viktigast, är att använda många olika k-mers i din montering. Du bör sedan utvärdera dina resultat och välja den bästa(s). Glöm aldrig att det nästan aldrig finns en enda korrekt montering.
- du bör noggrant hantera fel läser, innan du använder en k-mer. Om du inte försiktigt tar bort felen kan resultaten skapa en oönskad utbuktning som komplicerar din montering. Öka tröskeln för den felfrekvens du använder under sekvensbeskärning. Du kan förlora några sekvenser, men de som förblir kommer att vara de finaste.
- du bör hantera noggrant DNA-upprepningar. Till exempel genererar Illumina-sekvensering en mycket stor mängd data. Försök först att montera en liten del av läsningarna och använd dem alla för att upptäcka skillnader. Repeterbara korta läsningar kan störa din monteringsprocess negativt.
- Känn dina data. Om du inte känner till storleken på ditt förväntade genom, mängden sekvenseringstäckning och antalet läsningar, är du mer benägen att välja det bästa k-värdet för att montera ditt genom. Du kan besöka k-Mer-rådgivare, som velvet advisor från Monash university för att få råd om vilket värde som verkar mer lämpligt.
att använda k-mers av olika längder och anpassa contigs hjälper också forskare att upptäcka mutationshastigheter och utvidga dess användning. Naturligtvis, manipulera de Bruijn grafer mot montering fördel är inte ett universalmedel. Det finns många saker att tänka på än en förenklad funktion för att montera genomet hos en levande organism. Detta är bara en introduktion av historien och hur biologer kan använda den mer effektivt.
- Compeau PE, Pevzner PA, Tesler G. (2011). Hur man ansöker de Bruijn grafer till genomet montering.Natur Bioteknik. 29(11):987–91.
- Aggarwala V, Voight BF. (2016). En utvidgad sekvenskontextmodell förklarar i stort sett variabilitet i polymorfismnivåer över det mänskliga genomet. NaturGenetik. 48(4): 349–55.

har detta hjälpt dig? Vänligen dela med ditt nätverk.