introduktion
Kruppel-liknande faktor 4 (KLF4) är en medlem av familjen av zink-finger transkriptionsfaktor,som uttrycks i olika mänskliga vävnader. Det är välkänt som en av de fyra faktorerna för induktionen till pluripotenta stamceller (iPSCs) (Ghaleb och Yang, 2017). KLF4 kan reglera flera viktiga biologiska processer såsom neuroinflammation, oxidativ stress, proliferation, differentiering och apoptos (Kaushik et al., 2010; Mamonkin et al., 2013; Zhang et al., 2015; Miao et al., 2017; Xu et al., 2017). Mängder av tidigare studier fokuserade på KLF4: s roll i cancerutveckling och progression (Karam et al., 2017; Yadav et al., 2018). KLF4 är en transkriptionsfaktor med dubbla funktioner, som kan utöva sin roll som en onkogen eller en tumörsuppressorgen beroende på cancertyp eller cancerstadium (Evans och Liu, 2008). Det kan aktivera eller hämma transkription av gener som är involverade i cellproliferation, differentiering och apoptos (Ding et al., 2015). KLF4 kan samarbeta med andra omprogrammeringsfaktorer för att omvandla de somatiska cellerna till IPSC och hämma differentieringen av stamceller (Takahashi och Yamanaka, 2006; van Schaijik et al., 2018). Detta ger terapeutiska utsikter för kärlsjukdomar, immunsjukdomar, anorexi och andra sjukdomar (Imbernon et al., 2014; Liu Y. et al., 2015; Murgai et al., 2017). Dessutom kan KLF4 spela en allmänt reglerande roll i centrala nervsystemet (CNS). Flera studier tyder på att KLF4 är kopplat till flera neurologiska störningar, inklusive Alzheimers sjukdom (AD), epilepsi, Parkinsons sjukdom, hydrocephalus och schizofreni (Qin et al., 2011; Xie et al., 2013; Han et al., 2015; Nishiguchi et al., 2015; Li L. et al., 2017).
AD är en av de vanligaste kroniska neurodegenerativa sjukdomarna, vilket leder till kognitiva och minnesförluster, olika psykiska symtom och beteendeavvikelse och progressiv demens är den vanligaste kliniska funktionen (Jiang et al., 2018). De nuvarande bekräftade patogena faktorerna för AD innefattar bildandet av senila plack inducerade av onormal amyloid-2010-avsättning och neurofibrillära trassel eller dystrofisk neurit inducerad av tau-ackumulering (Querfurth och LaFerla, 2010; Shinohara et al., 2014). Dessutom kan AD också påverkas av genetiska faktorer. Emellertid är framkallningspatogenesen fortfarande Obskyr. De vanligaste läkemedlen för AD-behandling inkluderar neurotransmittorförstärkare, Anti-Amyloidmedel, neuroprotektiva peptider och andra läkemedel (Cacabelos, 2018). I synnerhet har flera studier visat att KLF4 spelade en viktig roll i patogenesen av AD. I den här översynen fokuserar vi på KLF4: s reglerande roll vid neuroinflammation, neuronal apoptos, axonal regenerering och järnackumulering för att förklara sambandet mellan KLF4 och patogenesen av AD, vilket kan ge insikter i de cellulära och molekylära mekanismerna för neurodegenerativa störningar.
de biologiska egenskaperna hos KLF4
KLF4 är ett zinkfingerinnehållande kärnprotein, isolerat från NIH 3T3-biblioteket och beläget i cellkärnan. Det identifierades först och kännetecknades av Shields et al. (1996). Molekylmassan för human KLF4 är 55kD och Den ligger på kromosomen 9q31. KLF4 täcker ett gensegment på 6,3 kb och har fem exoner. Dess cDNA-kodande region kodar för en polypeptid bestående av 470 aminosyrarester (Yet et al., 1998; Ghaleb och Yang, 2017). Karboxiterminalen för KLF4 har en DNA-bindningsstrukturregion innehållande tre zinkfingerstrukturer av Cys2His2 (C2H2) typ, som bildas av 81 mycket konserverade aminosyror. Det reglerar transkription med hög affinitet med CACCC-element och GC-rika målgen-DNA-sekvenser (Shields och Yang, 1998; Pearson et al., 2008). De flesta av DNA-bindningsställena för KLF4 är belägna inom zinkfingerregionen, inklusive N-terminal transkriptionsaktiveringsdomän för proteiner som interagerar, C-terminal zinkfingerstruktur för DNA-bindning och transkriptionshämningszon (Bieker, 2001). KLF4 är involverad i att reglera uttrycket av många endogena gener (Shields och Yang, 1998). Det finns en mycket variabel transkriptionsreglerande domän vid aminoterminalen för KLF4. Aminosyraresterna som ligger mellan 91 och 117-amino utgör en transkriptionell aktiveringsdomän, som är rik på prolin och serin, medan en transkriptionell förtrycksdomän också existerar. Därför har KLF4 två negativa effekter: aktivering och inhibering av gentranskription (Yet et al., 1998; Wei et al., 2006).
under den embryonala utvecklingen var KLF4 högre uttryckt i det sena stadiet av embryonal utveckling. I mogna vävnader och organ uttrycks KLF4 huvudsakligen i mag-tarmkanalen, munhålan, hudens epidermis, vaskulärt endotel och njure och uttrycks mindre i hjärnan (Segre et al., 1999; Ghaleb et al., 2011; Liu et al., 2013; Chen et al., 2015; Han et al., 2015; Bin et al., 2016). Det tros spela en viktig roll för att reglera cellproliferation och differentiering. Dessutom kan KLF4 också reglera cellcykeln. KLF4 kan aktivera P21 på ett P53-beroende sätt (Zhang et al., 2000). Dessutom fann man att KLF4 ( – / – ) celler gick in i senescensfasen tidigare än KLF4 ( + / + ) celler, vilket kan förklaras av det mindre antioxidantgenuttrycket och högre reaktiva syrearter (ROS) – nivå i KLF4 ( – / – ) celler. ROS kan öka p53-och p21-uttrycket och därefter främja DNA-skadan (Liu C. et al., 2015). Det visade sig att PRMT5 kan höja KLF4-uttrycket i proteinnivåer. PRMT5 rapporterades öka transkriptionen av p21 och minska uttrycket av bax via inhibering av KLF4 ubiquitylation (Hu et al., 2015). Dessutom har många studier visat att KLF4 är involverad i reglering av apoptos av neuroner (Kong et al., 2016; Cui et al., 2017; sång et al., 2018). Fysiologisk reglerande roll för KLF4 som vi har känt är fortfarande liten och ytterligare undersökningar behövs.
Roll KLF4 i AD
det är väl etablerat att AD huvudsakligen kännetecknas av minnes-och kognitiva funktionsnedsättningar och verkställande dysfunktion (Goedert och Spillantini, 2006). Många studier har visat att neuronal apoptos och synaptisk dysfunktion är patologisk grund för minskningen av kognitiv funktion (Caccamo et al., 2017; Guo et al., 2017; Yoon et al., 2018). Den ackumulerade skadan av en avsättning av en S. K. A., oxidativ stress och järnackumulering kan leda till neuronal dysfunktion och apoptos hos AD-patienter. Flera studier har visat att KLF4: s regulatoriska roll verkar vara avgörande i CNS. Med tanke på att KLF4 rapporterades reglera neuronal apoptos, synaptisk regenerering, oxidativ stress och neuroinflammation, kan förhållandet mellan KLF4 och patogenesen av AD vara ett potentiellt nytt mål för AD-behandling.
KLF4: s roll i Neuroinflammation
mängder av kliniska studier har visat att en Macau kan aggregera och är huvudkomponenten i de extracellulära avsättningarna i hjärnvävnaden hos AD-patienter, vilket kan försämra de omgivande synapserna och neuronerna och leda till neuronal död. Onormal sekretion eller överdriven produktion av en exporterande tillverkare leder till patologiska förändringar av AD, så en deponering av en exporterande tillverkare är Kärnlänken för AD (Rajmohan och Reddy, 2017). Dessutom har studier visat att överdriven a-avsättning kan stimulera gliaceller att utsöndra ROS och andra påverkande faktorer, vilket leder till oxidativ stress. Det var känt att oxidativ stress kan stimulera produktionen av en GHz. Därför kan en bisexuell och oxidativ stress interagera med varandra och påverka utvecklingen av AD (Cheignon et al., 2018).
KLF4 rapporterades som en potentiell modulator och har stor effekt på inflammation genom att förmedla makrofager och endotelceller (Figur 1) (Yoshida et al., 2014; Kapoor et al., 2015; Yang et al., 2018). I CNS kan överdrivna och kroniska inflammatoriska reaktioner orsaka skador på neuron och neurogliocyt. Det demonstrerades nyligen att KLF4-uttrycket positivt korrelerade med en Autoimmun42-inducerad neuroinflammation. I mikrogliala BV2-celler kan oligomeriskt a-Värde42 öka KLF4-uttrycket, vilket medieras av aktiverad P53 (Li L. et al., 2017). Under inflammatoriska betingelser, såsom en ackumulation av ax, kan frisättningen av proinflammatoriska cytokiner stimuleras vid genereringen av AD (Griffin och Barger, 2010). Neurotoxicitetspotens och proinflammatorisk potens av lösliga a-oligomerer i A. 42 är relativt högre än olöslig fiberavsättning (Selkoe, 1991; Weinberg et al., 2018). Silence of KLF4 kan återställa en GHz 42-medierad neuroinflammation, och överuttryck av KLF4 kan förvärra en 42-medierad Neuroinflammation (Li L. et al., 2017). En ackumulation av accumulation inducerar aktivering av astrocyter och mikroglia (Rodr Uscuguez et al., 2016). Aktiverade astrocyter kan förbättra neuroinflammationen genom att frigöra proinflammatoriska faktorer som IL-1, IL-6 och TNF-exporterande tillverkare (Rubio-Perez och Morillas-Ruiz, 2012; dom Actuicl et al., 2016). Den onda cykeln av inflammatoriska svar leder så småningom till dysfunktion och neuronal apoptos.
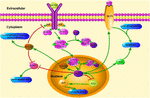
FIGUR 1. Schematillustration av KLF4 – relaterade signalvägar. Denna siffra belyser KLF4: s roll i neuroprotektion och axonregenerering. Pilarna i figuren indikerar aktivering eller marknadsföring, och de raka linjerna indikerar relaterad hämning. KLF4, Kruppel-liknande faktor 4; STAT3, signalomvandlare och aktivator av transkription 3; JAK, Janus Kinas; SOCS3, Suppressor av cytokinsignalering 3; HCP1, hembärarprotein 1; ERK5, mitogenaktiverat proteinkinas 5.
KLF4 spelar en avgörande roll för att reglera proinflammatoriska signaler. I gliaceller ökar gemfibrozil-inducerad KLF4-aktivering suppressor av cytokinsignal 3 (SOCS3) via PI3-Kinas-AKT-vägen (Ghosh och Pahan, 2012). Den SiRNA-medierade knockdownen av KLF4 kunde dämpa nivån av SOCS i astroglia och microglia hos möss, vilket senare kan påverka uttrycket av inflammatorisk gen (Kaushik et al., 2010; Ghosh och Pahan, 2012). Dessutom kan SoCs-radering främja överlevnaden av skadade neuroner och främja axonregenerering (Smith et al., 2009; Sun et al., 2011). Och KLF4 reglerar positivt produktionen av IL-1 msk eller andra proinflammatoriska markörer. Det reglerar positivt cyklooxygenas-2 (Cox-2) och reglerar negativt inducerbart kväveoxidsyntas (iNOS) (Kaushik et al., 2013). Dessutom är KLF4 en viktig regleringsfaktor för monocytdifferentiering och ett potentiellt mål för immunreglering (Alder et al., 2008). Därför kan KLF4 främja neuroinflammation genom att reglera dessa negativa regulatorer.
det är värt att nämna att KLF4 i Parkinsons sjukdomsmodell kan främja MPP+-inducerad oxidativ stress och neurotoxicitet och sedan öka neuronal apoptos och fördröja cellproliferationen (Chen et al., 2013). Oxidativ stress är en obalans mellan peroxidation och antioxidation. Fria radikaler kan orsaka förändringar i olika makromolekyler, vilket leder till cellskador, cellåldring och vävnadsskada (Parajuli et al., 2013; Nie et al., 2015). Oxidativ stress kan förvärra tidig inflammation och en produktionen av en produktion av ax och sedan förvärra AD (Cai et al., 2011). Därför kan KLF4 vara involverad i oxidativ stress i AD.
dessa fynd antyder KLF4 en nyckelroll för att förmedla neuroinflammation genom att aktivera mikroglia och följaktligen frisättning av proinflammatoriska cytokiner. Det har potential att förbättra neuroinflammation. Hittills har många studier om patogenesen av AD fokuserat på neuroinflammation. Som ett potentiellt mål för immunreglering kan KLF4 främja de inflammatoriska svaren hos mikroglia via påverkande relaterade negativa regulatorer, vilket har stor effekt på utvecklingen av AD.
Roll KLF4 i apoptos
neurodegenerativa förändringar inkluderar gradvis förlust av neuroner och synapser i de representativa hjärnregionerna, såsom hjärnbarken, hippocampus och andra subkortiska regioner. Funktionsnedsättningarna av CNS inducerad av neuronal förlust är permanenta (Citron, 2010). Ihållande oxidativ stress kan leda till neuronal apoptos (Wu et al., 2010). Ett stort antal studier har bekräftat att AD är nära relaterat till oxidativ stress (Lee et al., 2012; Yui et al., 2015). Det visade sig att kronisk oxidativ stress kan förbättra uttrycket av fosfolipas A2-grupp 3 (Pla2g3) i astrocyter och störa balansen i en AA, och följaktligen leda till utvecklingen av AD (Yui et al., 2015).
många studier har visat att KLF4 spelar en viktig roll för att hämma utvecklingen av oxidativ stress (Shi et al., 2014; Liu C. et al., 2015). Det visade sig att KLF4 kan främja cellerna apoptos inducerad av H2O2, denna åtgärd kommer sannolikt att orsakas av ökat bax-uttryck och minskat Bcl-2-uttryck (Li et al., 2010). Quercetin kan minska KLF4-uttrycket i humana neuroblastom SH-SY5Y-celler och öka uttrycket av BCL-2/bax-förhållandet. Vidare kan Quercetin moderera apoptoshastigheten för SH-5YSY-cellen och minska kaspas-3-enzymaktiviteten (Xi et al., 2012). En ny studie undersökte den neuroprotektiva effekten av mitogenaktiverat protein (MAP) Kinas 5 (ERK5) mot oxidativ stress. Aktivering av ERK5 kan delvis minska H2O2-inducerad hippocampal neurons död och öka NGF – och PC-inducerad neuroprotektion (Su et al., 2014). Nils et al. använde en mutant av MEK5 (MEK5D) för att studera ERK5-aktiverad transkription och funktionella svar i humana endotelceller och identifierade KLF4 var ett nytt nedströms erk5-mål (Ohnesorge et al., 2010). Det visade sig att överuttryck av KLF4 kan undertrycka TNF-medierade inflammatoriska svar och minska leukocytadhesion och basalcellapoptos. Dessa resultat bekräftar att KLF4 har antiinflammatoriska och anti-apoptotiska egenskaper (Ohnesorge et al., 2010). Efterföljande experiment har visat att försvinnandet av cerebral cavernös missbildning 1 (CCM1) i endotelceller aktiverar erk5 via MEKK3-MEK5 signalväg och ökar KLF4-uttrycket (Cuttano et al., 2016). ERK5 spelar en medierande roll i förkonditionering (PC) och nervtillväxtfaktor (NGF) uppreglerade uttrycket av KLF4 (Su et al., 2014). Dessutom kan RNAi-medierad nedslagning av KLF4 också minska NGF-eller PC-inducerad neuroprotektion. Överuttryck av KLF4 leder till högre Bcl-2 / bax-förhållande i H2O2-stressade celler (Su et al., 2014). Överuttryckt KLF4 accelererar förändringar i bcl-2 och bax genom att kombinera med motsvarande promotor (Li et al., 2010). Erk5 / KLF4-kaskad kan fungera som en pivot i olika vägar som skyddar neuroner från oxidativ stressinducerad död (Su et al., 2014).
oxidativ stress har ansetts vara nära relaterad till många degenerativa sjukdomar. KLF4 spelar viktiga roller för att upprätthålla genomisk stabilitet vid oxidativ stress. KLF4 och ERK5 verkar tillsammans för att skydda neuroner från oxidativ stressinducerad apoptos. Därför kan KLF4 fungera som ett terapeutiskt mål för att agera mot oxidativ stress när den aktiveras. Det har rapporterats att statinläkemedel kan aktivera ERK5, vilket leder till uttryck av KLF4 och dess beroende gener (Ohnesorge et al., 2010), men mekanismen förblir oklar, och KLF4-relaterade uppströms-och nedströms målgener studeras mindre i oxidativ stress, det finns ett behov av ytterligare studier.
Roll KLF4 i Axonregenerering
tidig axonförlust är ett vanligt inslag i neurodegenerativa sjukdomar. Synaptisk förlust och transportstörning i AD kan orsaka kognitiva funktionsnedsättningar (Holtzman et al., 2011; Coleman, 2013). Graden av deklarativ minnesskada är relaterad till den synaptiska densiteten i hippocampus och cortex. Lösliga a-oligomerer av A-typ minskar glutamatupptaget och främjar synaptisk dysfunktion, vilket stör synaptisk plasticitet (Li et al., 2009). Därför är det särskilt viktigt att studera hur man reparerar axonerna i CNS. I retinala ganglionceller har axoner en stark förmåga att växa och regenerera under tidig utveckling, men i CNS hos vuxna däggdjur förlorar axoner sin regenereringskapacitet och neuronerna kan ta examen för att dö eller atrofi (Goldberg och Barres, 2000; Goldberg et al., 2002).
KLF4 spelar en viktig roll för att hämma axontillväxt. I embryonala RGCs kan överuttryck av KLF4 minska andelen neuritförlängning, längden på axoner och dendriter och neuritförgreningen. Dessutom fann man att ÖVERUTTRYCKET av KLF4 kan minska långsiktiga postnatala axontillväxthastigheter men misslyckades med att minska kortsiktiga axontillväxthastigheter (Moore et al., 2009; Steketee et al., 2014). Senare studier har funnit att axonbuntarna av KLF4-cKO-möss var tjockare än kontrollmöss (Fang et al., 2016). Dessutom kan avlägsnande av KLF4-uttryck under utveckling öka reproduktionspotentialen hos vuxna RGCs. Dessutom hade KLF4 som saknade den c-terminala DNA-bindningsdomänen ingen effekt på axontillväxten. Det fanns ingen inverkan på cellernas överlevnad efter att retinala ganglionceller skadades om KLF4 slog ut (Moore et al., 2009).
KLF4 kan också påverka axonal regenerering. En ny studie rapporterade att minskningen av KLF4-uttryck i vuxna retinala ganglionceller främjade axonregenerering genom jak-STAT3-vägen (Qin et al., 2013). KLF4 ökade fosforyleringen av STAT3 och reglerade axontillväxten via JAK-STAT-signalering (Qin och Zhang, 2012). Under behandlingen av cytokiner fosforyleras medlemmar av statfamiljen av proteiner vid de karboxiterminala tyrosin-och serinställena i cellen för att bilda en stabil dimer. Denna modifiering förbättrar transkriptionen av cellassocierade gener (Yuan et al., 2005). Interaktionen mellan KLF4 och STAT3 på cytokininducerad fosforylering av tyrosin705 hämmar uttrycket av STAT3 genom att hämma bindningen av STAT3 till DNA (Qin et al., 2013). KLF4 knockdown förbättrar uppenbarligen axons regenerering i retinala ganglionceller efter skada av optisk nerv och förhindrar nerven från skada efter mild hjärnskada. Åtgärderna förmedlas av en minskning av p-p53 och en ökning av pstat3-nivåerna. KLF4 reglerar positivt neuronal apoptos via p53-och JAK-STAT3-vägarna, och KLF4 reglerar negativt axonal reparation via JAK-STAT3-vägen (Cui et al., 2017).
därför antog vi att i AD kan axonal regenerering åstadkommas genom att ändra uttrycket av KLF4 eller ändra intracellulära relaterade signalvägar och kontrollera ad-progression genom att minska saknade axoner eller minska axonal dysfunktion. Men hur man använder KLF4-transkriptionsfaktorn i potentiell terapi behöver fortfarande ytterligare utforskning.
roll av KLF4 i järnackumulering
järn finns allmänt i biologiska system, järnrelaterade metalloproteinaser spelar en nyckelroll vid transport av syre, överföring av elektroner och katalysering av biokemiska reaktioner (Aisen et al., 2001). Eventuellt överskott av järn utöver det normala fysiologiska området kan dock skada människors hälsa (Adlard och Bush, 2006). Studier har funnit att järninnehållet i hippocampus är negativt korrelerat med utförandet av minnestester (Ding et al., 2009). Ökad järnbelastning i hjärnan accelererar bildandet av en 2a-plack och hyperfosforylerade tau-trassel, samtidigt som oxidativ stress förbättras (Peters et al., 2015). Järn, som har en hög grad av permeabilitet, främjar nervtillväxt och cell-till-cell-anslutningar under hjärnans utveckling (Dallman och Spirito, 1977).
en ny studie visade att fysiologisk stress orsakade aktivering av KLF4-HCP1-signalvägen och ökat hemupptag (Li H. et al., 2017). Heme står för 95% av det funktionella järnet i människokroppen. Det är en av huvudkomponenterna i heme-oxygenas (Hooda et al., 2014; Kurucz et al., 2018). Att öka aktiviteten av oxygenas – 1 kan fördröja oxidationen av den åldrande hjärnan (Verdile et al., 2015; Serini och Calviello, 2016; Kurucz et al., 2018). Detta har en lättnadseffekt på AD. Fysiologisk stress inducerar glukokortikoidnivåhöjning, glukokortikoid ökar hembärarproteinet 1 (HCP1) uttryck via KLF4, och sedan främjar HCP1 hemupptag (Li H. et al., 2017). Glukokortikoid och KLF4 reglerar antiinflammatoriska gener tillsammans, och celler med lågt glukokortikoidinnehåll kan inte helt inducera KLF4-uttryck (Sevilla et al., 2015). KLF4-inducerad ökning av hemintag leder till järnackumulering i hjärnan. Järn främjar frisättningen av ROS (Tronel et al., 2013). Järnelement förbättrar hjärnans oxidativa stress hos råttor under psykisk stress (Yu et al., 2011). Därför kan HCP1 regleras av KLF4 och glukokortikoid tillsammans. Ökande HCP1 förbättrar hemupptaget, vilket leder direkt till järnackumulering i hjärnan, förvärrar oxidationen, ökar apoptos eller dysfunktion och förvärrar hjärnskador.
det är allmänt accepterat att minnes-och inlärningssvårigheter är de viktigaste symptomen på AD. Ett stort antal kliniska data har visat att en plack belastning och järn ackumulering svar på utvecklingen av lärande och kognitiv dysfunktion i AD (van Bergen et al., 2018). Nyligen publicerade data har antytt att högdosjärn ökar en avsättning av hCG och dämpar inlärning och minne hos möss (Guo et al., 2013). Kliniska studier har visat att järninnehållande mikroglia finns i hippocampus hos AD-patienter under magnetisk resonansavbildning (Zeineh et al., 2015). Microglia förvärvar järn från överföring eller icke-överföring, extracellulära och intracellulära källor (McCarthy et al., 2018). Selektivt och ihållande KLF4-uttryck kan induceras i kärnan och cytoplasman hos ischemiska hippocampala reaktiva astrocyter (Park et al., 2014). Studier har visat att KLF4 fungerar som en transkriptionell repressor. Det reglerar uttrycket av älg-3, och sedan hämmar älg-3 uttrycket av HO-1 (Tsoyi et al., 2015). Heme oxygenase – 1 (HO-1) är ett stressprotein som bryter ner heme till bilirubin, fritt järn och kolmonoxid. Uppreglering av HO – 1 i astrocyter kan leda till onormal järnavsättning och mitokondriell dysfunktion i hjärnan, vilket leder till minskad kognitiv förmåga (Schipper, 1999, 2004). Därför kan KLF4 vara involverad i processen med järnackumulering i astrocyter, vilket förvärrar oxidation i AD och förvärrar hjärnskador.
slutsats
KLF4 är allmänt känt för att spela en central roll vid reglering av cellproliferation, apoptos och differentiering. Tidigare studier har fokuserat på reglering av KLF4 i flera viktiga neurofysiologiska processer, inklusive neuroinflammation, neuroprotektion och synaptisk regenerering. Nyligen har KLF4 visat sig spela en viktig roll i patogenesen av AD. I den här artikeln granskar vi KLF4: s roll i neuroprotektion och neurogenes i AD.
KLF4 är inte bara en regulator för reglering av cellproliferation och differentiering utan också ett potentiellt mål för reglering av immunsvar. KLF4 kan reglera negativa inflammatoriska faktorer och främja inflammatoriskt svar och ha stor effekt på uttrycket av astrocytkärnmikroglia. Dessutom kan KLF4 och ERK5 agera tillsammans för att utöva neuroprotektiva åtgärder. Vidare kan axonregenerering åstadkommas genom att ändra innehållet i specifika transkriptionsfaktorer, intracellulära hämmare eller förändra intracellulära signalvägar. Att slå ut KLF4 kan förbättra axonregenerationen och påskynda axontillväxten. Reduktion av KLF4-uttryck främjar axonregenerering genom jak-STAT3-vägen, och KLF4 främjar JAK-STAT3-vägen till ytterligare axonregenerering. Därför kan KLF4 vara involverad i processen med antiinflammatorisk, anti-apoptos, axonregenerering och järnackumulering i CNS, som spelar en central roll i AD-generationen. Dessa fynd tyder på att KLF4 representerar ett potentiellt terapeutiskt mål för AD. De djupa cellulära och molekylära mekanismerna för effekterna av KLF4 på AD förblir emellertid oklara och ytterligare undersökningar behövs.
Författarbidrag
ZQC, XHZ och YJ skrev manuskriptet. SHG och JYL modifierade ramen för manuskriptet. BJL och RJC tillhandahöll de kritiska revideringarna. Alla författare godkände den slutliga versionen av manuskriptet för inlämning.
finansiering
detta arbete stöddes av bidrag från Natural Science Foundation of China (NSFC) (81871070, 31571126, 31471120) och Jilin Science and Technology Agency-finansiering (bidrag nr 20180519003JH, 20180414051GH, 20170414034GH och 20180414050gh).
intressekonflikt uttalande
författarna förklarar att forskningen genomfördes i avsaknad av kommersiella eller finansiella relationer som kan tolkas som en potentiell intressekonflikt.
Cacabelos, R. (2018). Har det skett förbättringar i Alzheimers sjukdom läkemedelsupptäckt under de senaste 5 åren. Exp. Opin. Drog Discov. 13, 523–538. doi: 10.1080/17460441.2018.1457645
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Liu, Y., Shi, J. och Chen, X. (2015). Identifiering av nya mål för säsongsallergisk rinit under och utanför pollensäsongen genom mikroarrayanalys. Acta Otolaryngol. 135, 1330–1336. doi: 10.3109 / 00016489.2015.1067906
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Serini, S. och Calviello, G. (2016). Reduktion av oxidativ / nitrosativ stress i hjärnan och dess engagemang i den neuroprotektiva effekten av n-3 PUFA vid Alzheimers sjukdom. Curr. Alzheimers Res. 13, 123-134. doi: 10.2174/1567205012666150921101147
PubMed Abstrakt / CrossRef fulltext / Google Scholar