teknologien FOR DNA-sekvensering ble utviklet tilbake i 1977 takket Være Frederick Sanger. Det tok litt lengre tid før det var mulig å sekvensere et komplett genom. Dette skyldes at vi trengte en passende matematisk modell og massiv beregningskraft for å samle millioner eller milliarder av små leser til et større komplett genom. Dagens datakraft og programvare er hovedforskjellen mellom det som pleide å ta mange års arbeid tidlig på 2000-tallet og det som bare tar bare noen få timer i dag. Algoritmen du valgte å gjøre dette er den» hellige gral » av monteringsteknologien. Disse algoritmene innlemme en av de mest kjente variablene kjent i matematiske modeller, k-mer.
opprinnelsen til k-mer og den matematiske modellen som omgir den kommer fra En 1735 Sveitsisk matematiker Leonhard Euler, som er kjent som den matematiske funksjonens far. En nederlandsk matematiker Nicolaas De Bruijn tilpasset eulers ideer for å finne en syklisk sekvens av bokstaver tatt fra et gitt alfabet som alle mulige ord av en viss lengde vises som en streng av påfølgende tegn i den sykliske sekvensen nøyaktig en gang.
de Bruijns algoritme ble tilpasset av molekylærbiologer, som mange år senere møtte et tilsvarende problem: hvordan man monterer DNA-sekvenser. Dermed bruker forskere over hele verden nå De Bruijn-grafen og variabelen k.
Anvendelse av k-mers For Å Sette SAMMEN DNA-Sekvenser
med noen få ord, de novo genome assembly innebærer å koble påfølgende små DNA-leser og ende opp med større sekvenser. For å generere En De Bruijn-Graf (se figuren nedenfor), må nukleotidene ved kanten av hver les overlappe kanten av en annen (og så videre). Det endelige målet er å skape et sammenhengende toppunkt, som (potensielt) vil resultere i store DNA-fragmenter.
du må fragmentere dine leser i k-mers, som er et bestemt antall nukleotider som overlapper. K-mer lar deg generere en unik sekvens fra mange små. Hver unike k-mer sekvens er identifisert og ekstra kopier er eliminert. Dette aspektet av k-mers lar deg overvinne en av ulempene ved neste generasjons sekvensering-å få leser som representerer genomiske regioner med forskjellige frekvenser (dvs.å få mange små leser fra en region). Bruken av k-mers eliminerer sekvenser gjentatt mer enn en gang på grunn av ulik sekvensdekning. Vær imidlertid oppmerksom på at en lav k-mer-størrelse vil øke sjansene for at nukleotider overlapper, mens de har en større verdi, vil redusere dem.
dagens de novo-monteringsteknologi er mer effektiv når du bruker biblioteker med store leser (dvs. 1.000–10.000 bps) kombinert med mindre (100-200 bps). Programmer kan bruke k verdi og k-mers å montere kort leser. Disse kan da bli innarbeidet og verifisert av større for å ende opp i mer nøyaktige contigs.
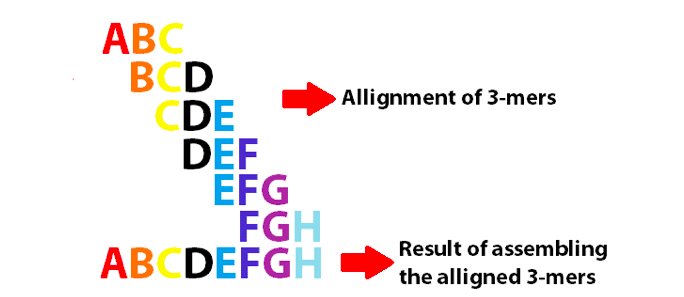
Eksempel på en De Bruijn-graf som bruker 3-mers til å sette sammen de 8 første bokstavene i det engelske alfabetet. Merk at disse 3-mers overlapper som k-1.
Jo Mer Du Vet, Desto Mer Kan DU Oppnå I DNA-Montering
det er spesifikke tips du må vurdere før du bruker De Bruijn-grafer i monteringsmetoden og velger den mest passende k-mer-størrelsen. Ved å utnytte disse, kan du generere bedre resultater.
- Først av alt, og kanskje viktigst, er å bruke mange forskjellige k-mere i forsamlingen. Du bør deretter vurdere resultatene dine og velge den beste (e). Glem aldri at det er nesten aldri en og bare en riktig montering.
- du bør nøye håndtere feil leser, før du bruker en k-mer. Hvis du ikke fjerner feilene forsiktig, kan resultatene skape en uønsket bulge, noe som kompliserer samlingen din. Øk terskelen for feilfrekvensen du bruker under sekvens trimming. Du kan miste noen sekvenser, men de som forblir vil være de fineste.
- du bør håndtere NØYE DNA-repetisjoner. For Eksempel genererer Illumina-sekvensering en meget stor mengde data. Prøv først å sette sammen en liten brøkdel av lesingene, og bruk dem alle til å oppdage forskjeller. Repeterbare korte leser kan forstyrre negativt med monteringsprosessen.
- Kjenn dine data. Hvis du ikke vet størrelsen på ditt forventede genom, mengden sekvenseringsdekning og antall leser, er du mer tilbøyelig til å velge den beste k-verdien for å montere genomet ditt. Du kan besøke k-mer advisors, som velvet advisor Fra Monash university for å få råd om hvilken verdi som synes mer egnet.
Ved hjelp av k-mers av ulike lengder og samkjøre contigs hjelper også forskere til å oppdage mutasjonsrater, utvide bruken. Selvfølgelig, manipulere De Bruijn grafer mot montering fordel er ikke et universalmiddel. Det er mange ting å vurdere enn en forenklet funksjon for å samle genomet til en levende organisme. Dette er bare en introduksjon av historien og hvordan biologer kan bruke den mer effektivt.
- Compeau PE, Pevzner PA, Tesler G. (2011). Hvordan søke De Bruijn grafer til genome assembly.Natur Bioteknologi. 29(11):987–91.
- Aggarwala V, Voight BF. (2016). En utvidet sekvens kontekst modell forklarer bredt variasjon i polymorfisme nivåer over det menneskelige genom. Naturgenetikk. 48(4): 349–55.

har dette hjulpet deg? Vennligst del med nettverket ditt.