Introduksjon
Kruppel-lignende faktor 4 (KLF4) er medlem av familien av sink-finger transkripsjonsfaktor,som uttrykkes i forskjellige humane vev. Det er kjent som en av de fire faktorene for induksjon til pluripotente stamceller (iPSCs) (Ghaleb Og Yang, 2017). KLF4 kan regulere flere viktige biologiske prosesser som nevroinflammasjon, oksidativt stress, proliferasjon, differensiering og apoptose (Kaushik et al., 2010; Jørgen et al., 2013; Zhang et al., 2015; Miao et al., 2017; Jørgen et al., 2017). Mengder av tidligere studier fokusert PÅ KLF4 rolle i kreft utvikling og progresjon (Karam et al., 2017; Yadav et al., 2018). KLF4 er en dual-funksjon transkripsjonsfaktor, som kan utøve sin rolle som et onkogen eller et tumor suppressorgen avhengig av krefttype eller kreftstadium (Evans Og Liu, 2008). Det kan aktivere eller hemme transkripsjon av gener involvert i celleproliferasjon, differensiering og apoptose (Ding et al., 2015). KLF4 kan samarbeide med andre omprogrammeringsfaktorer for å konvertere de somatiske cellene til iPSCs og hemme differensiering av stamceller (Takahashi og Yamanaka, 2006; van Schaijik et al., 2018). Dette gir terapeutiske utsikter for vaskulære sykdommer, immunforsvar, anoreksi og andre sykdommer (Imbernon et al., 2014; Liu Y. et al., 2015; Murgai et al., 2017). VIDERE KAN KLF4 spille en mye regulerende rolle i sentralnervesystemet (CNS). Flere studier indikerer AT KLF4 er knyttet til flere nevrologiske lidelser, inkludert Alzheimers sykdom (AD), epilepsi, Parkinsons sykdom, hydrocephalus og schizofreni (Qin et al., 2011; Xie et al., 2013; Han et al., 2015; Nishiguchi et al., 2015; Li L. et al., 2017).
AD er en av de vanligste kroniske nevrodegenerative sykdommene, noe som fører til kognitive og hukommelsessvikt, ulike mentale symptomer og atferdsavvik og progressiv demens er den vanligste kliniske funksjonen (Jiang et al ., 2018). De nåværende bekreftede patogene faktorene for AD inkluderer dannelse av senile plaques indusert av unormal amyloid-β (Aß) avsetning og nevrofibrillære tangles eller dystrofisk nevitt indusert av tau akkumulering (Querfurth og LaFerla, 2010; Shinohara et al., 2014). I TILLEGG KAN AD også påvirkes av genetiske faktorer. Imidlertid er den fremkallende patogenesen fortsatt uklar. De mest utbredte stoffene for AD-behandling inkluderer nevrotransmitterforsterkere, anti-Amyloidmidler, nevrobeskyttende peptider og andre stoffer (Cacabelos, 2018). Spesielt har flere studier vist AT KLF4 spilte en betydelig rolle i patogenesen AV AD. I denne gjennomgangen, vi fokuserer på regulatoriske rolle KLF4 i nevroinflammasjon, neuronal apoptose, aksonal regenerering, og jernakkumulering for å forklare sammenhengen MELLOM KLF4 og patogenesen AV AD, som kan gi innsikt i de cellulære og molekylære mekanismer av nevrodegenerative lidelser.
De Biologiske Egenskapene TIL KLF4
KLF4 er et sinkfingerholdig kjerneprotein, isolert FRA NIH 3T3-biblioteket og lokalisert i cellekjernen. Det ble først identifisert og preget Av Shields et al. (1996). Den molekylære massen av menneskelig KLF4 er 55kD og den ligger på kromosomet 9q31. KLF4 dekker et 6,3 kb gensegment og har fem eksoner. Dens cDNA-kodende region koder for et polypeptid bestående av 470 aminosyrerester (Ennå et al.( 1998; Ghaleb Og Yang, 2017). Karboksyterminalen TIL KLF4 har EN DNA-bindende strukturregion som inneholder Tre cys2his2 (C2H2) type sinkfingerstrukturer, som dannes av 81 høyt konserverte aminosyrer. Det regulerer transkripsjon av høy affinitet MED CACCC elementer OG GC-rike målet genet DNA-sekvenser (Shields Og Yang, 1998; Pearson et al., 2008). De FLESTE AV DNA-bindende steder AV KLF4 er lokalisert innenfor sink finger regionen, inkludert N-terminal transkripsjon aktivering domene for proteiner interaksjon, C-terminal sink finger struktur FOR DNA binding og transkripsjon hemming sone (Bieker, 2001). KLF4 er involvert i å regulere uttrykket for mange endogene gener(Shields And Yang, 1998). Det er et svært variabelt transkripsjonsregulatorisk domene ved aminoterminalen TIL KLF4. Aminosyrerester som ligger mellom 91 og 117-aminoen utgjør et transkripsjonelt aktiveringsdomene, som er rik på prolin og serin, mens et transkripsjonelt undertrykkelsesdomene også eksisterer. DERFOR HAR KLF4 to bivirkninger: aktivering og inhibering av gentranskripsjon (Ennå et al., 1998; Wei et al., 2006).
UNDER embryonal utvikling var KLF4 høyere uttrykt i sent stadium av embryonal utvikling. MENS i modne vev OG organer, er KLF4 hovedsakelig uttrykt i mage-tarmkanalen, munnhulen, hud epidermis, vaskulær endotel og nyre, og er mindre uttrykt i hjernen (Segre et al ., 1999; Ghaleb et al., 2011; Liu et al., 2013; Chen et al., 2015; Han et al., 2015; Jørgen et al., 2016). Det antas å spille en betydelig rolle i å regulere celleproliferasjon og differensiering. DESSUTEN KAN KLF4 også regulere cellesyklus. KLF4 kan aktivere P21 På En p53-avhengig måte (Zhang et al., 2000). I tillegg ble DET funnet AT KLF4 ( – / – ) celler gikk inn i senescensfase tidligere ENN KLF4 ( + / + ) celler, som kan forklares av det mindre antioksidantgenuttrykket og høyere reaktive oksygenarter (ROS) nivå I KLF4 ( – / – ) celler. ROS kan øke p53 og p21 uttrykk og deretter fremme DNA-skade (Liu C. et al., 2015). DET ble funnet AT PRMT5 kan heve KLF4-uttrykket i proteinnivåer. PRMT5 ble rapportert å øke transkripsjonen av p21 og redusere ekspresjonen av bax via inhibering AV klf4 ubiquitylering (Hu et al., 2015). Videre har mange studier vist AT KLF4 er involvert i regulering av apoptose av nevroner (Kong et al., 2016; Cui et al., 2017; Song et al., 2018). Fysiologisk regulerende rolle KLF4 som vi har kjent er fortsatt lite og videre undersøkelser er nødvendig.
Rolle KLF4 I AD
DET er vel etablert at AD er hovedsakelig preget av hukommelse og kognitive svekkelser og utøvende dysfunksjon (Goedert Og Spillantini, 2006). Mange studier har vist at neuronal apoptose og synaptisk dysfunksjon er patologisk grunnlag for nedgangen i kognitiv funksjon (Caccamo et al., 2017; Guo et al., 2017; Jørgen et al., 2018). Akkumulert skade på Enß avsetning, oksidativt stress og jernakkumulering kan føre til neuronal dysfunksjon og apoptose hos AD-pasienter. Flere studier har vist AT KLF4S regulatoriske rolle ser ut til å være avgjørende for CNS. Tatt I betraktning AT KLF4 ble rapportert å regulere nevronal apoptose, synaptisk regenerering, oksidativt stress og nevroinflammasjon, kan forholdet MELLOM KLF4 og patogenesen av ALZHEIMERS sykdom være et potensielt nytt mål for ALZHEIMERS behandling.
Rolle KLF4 I Nevroinflammasjon
Mengder av kliniske studier har vist at Enß kan aggregere og er hovedkomponenten i de ekstracellulære forekomster av hjernevevet AV AD pasienter, som kan svekke de omkringliggende synapser og nevroner, og føre til neuronal død. Unormal sekresjon eller overdreven produksjon av Enß fører til patologiske endringer AV AD, så Enß deponering er kjernekoblingen TIL AD (Rajmohan and Reddy, 2017). I tillegg har studier vist at overdreven Enß avsetning kan stimulere glialceller til å utskille ROS og andre påvirkningsfaktorer, noe som fører til oksidativt stress. Det var kjent at oksidativt stress kan stimulere produksjonen av Enß. Derfor Kan Etß og oksidativt stress samhandle med hverandre og påvirke UTVIKLINGEN AV AD (Cheignon et al., 2018).
KLF4 ble rapportert som en potensiell modulator og har stor effekt på betennelse ved mediering av makrofager og endotelceller (Figur 1) (Yoshida et al ., 2014; Kapoor et al., 2015; Jan et al., 2018). I CNS kan overdreven og kronisk inflammatorisk reaksjon forårsake skade på nevron og neurogliocyt. DET ble nylig demonstrert at KLF4-uttrykket positivt korrelerte med Enß 42-indusert nevroinflammasjon. I mikrogliale bv2-celler kan oligomere Aß 42 øke klf4-uttrykket, som medieres av aktivert P53 (Li L. et al., 2017). Under betennelsestilstander, som For eksempel Akkumulering Av Enß, kan frigivelsen av proinflammatoriske cytokiner stimuleres ved GENERERING AV AD (Griffin og Barger, 2010). Nevrotoksisitet potens og pro-inflammatorisk potens av løselig Enß 42 oligomerer er relativt høyere enn uoppløselig fiber innskudd (Selkoe, 1991; Weinberg et al., 2018). Silence OF KLF4 er i stand til å gjenopprette Enß42-mediert nevrobetennelse, og OVERUTTRYKK AV KLF4 kan forverre Enß42-mediert nevrobetennelse (Li L. et al., 2017). Enß akkumulering induserer aktivering av astrocytter og microglia (Rodr ④guez et al., 2016). Aktiverte astrocytter kan forbedre nevroinflammasjonen ved å frigjøre proinflammatoriske faktorer SOM IL-1, IL-6 og tnf-α(Rubio-Perez og Morillas-Ruiz, 2012; Dom@né Et al., 2016). Den onde syklusen av inflammatoriske responser fører til slutt til dysfunksjon og neuronal apoptose.
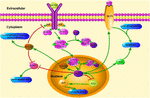
FIGUR 1. Schematicillustration AV KLF4 relaterte signalveier. Denne figuren fremhever KLF4S rolle i nevrobeskyttelse og axonregenerering. Pilene i figuren indikerer aktivering eller forfremmelse, og de rette linjene indikerer relatert inhibering. KLF4, Kruppel-lignende faktor 4; STAT3, Signaltransduser og aktivator av transkripsjon 3; JAK, Janus Kinase; SOCS3, Suppressor av cytokinsignalering 3; HCP1, hemebærerprotein 1; ERK5, mitogenaktivert proteinkinase 5.
KLF4 spiller en avgjørende rolle i å regulere pro-inflammatoriske signaler. I glialceller øker gemfibrozil-indusert KLF4-aktivering suppressor av cytokin signal 3 (SOCS3) via PI3-kinase-AKT-bane (Ghosh og Pahan, 2012). Den SiRNA-medierte knockdown AV KLF4 kunne dempe NIVÅET AV SOCS i astroglia og microglia av mus, som senere kunne påvirke uttrykket av inflammatorisk gen (Kaushik et al.(2010; Ghosh Og Pahan, 2012). I tillegg KAN SOCS-sletting fremme overlevelsen av skadede nevroner og fremme axonregenerering (Smith et al., 2009; Sol et al., 2011). OG KLF4 regulerer positivt produksjonen AV IL-1Β eller andre pro-inflammatoriske markører. Det regulerer positivt cyklooksygenase-2 (Cox-2) og regulerer negativt inducerbart nitrogenoksydsyntase (iNOS) (Kaushik et al., 2013). I TILLEGG ER KLF4 en viktig regulerende faktor for monocyttdifferensiering og et potensielt mål for immunregulering (Alder et al., 2008). DERFOR KAN KLF4 fremme nevroinflammasjon ved å regulere disse negative regulatorene.
DET er verdt å nevne AT I Parkinsons sykdomsmodell KAN KLF4 fremme MPP + – indusert oksidativt stress og nevrotoksisitet, og deretter øke neuronal apoptose og forsinke celleproliferasjonen (Chen et al., 2013). Oksidativt stress er en ubalanse mellom peroksidasjon og antioxidasjon. Frie radikaler kan forårsake endringer i forskjellige makromolekyler, noe som fører til celleskader, cellealdring og vevskader (Parajuli et al., 2013; Nie et al., 2015). Oksidativt stress kan forverre tidlig betennelse og Enß produksjon og deretter forverre AD (Cai et al., 2011). DERFOR KAN KLF4 være involvert i oksidativt stress i AD.
disse funnene antyder AT KLF4 har en nøkkelrolle i å megle nevroinflammasjon ved å aktivere microglia og dermed frigjøre proinflammatoriske cytokiner. Det har potensial til å forbedre nevroinflammasjon. Så langt har mange studier på patogenesen AV AD fokusert på nevrobetennelse. SOM et potensielt mål for immunregulering kan KLF4 fremme de inflammatoriske responsene til microglia via å påvirke relaterte negative regulatorer, noe som har stor effekt på utviklingen av AD.
Klf4s Rolle I Apoptose
Nevrodegenerative endringer inkluderer gradvis tap av nevroner og synapser i de representative hjernegruppene, som hjernebarken, hippocampus og andre subkortiske regioner. Funksjonsnedsettelser av CNS indusert av nevronetap er permanente (Citron, 2010). Vedvarende oksidativt stress kan føre til neuronal apoptose (Wu et al., 2010). Et stort antall studier har bekreftet AT AD er nært knyttet til oksidativt stress (Lee et al., 2012; Yui et al., 2015). Det ble funnet at kronisk oksidativt stress kan forbedre ekspresjonen Av Fosfolipase a2 gruppe 3 (Pla2g3) i astrocytter og forstyrre balansen Av Enß, og dermed føre til utvikling AV AD (Yui et al., 2015).
Mange studier har vist AT KLF4 spiller en viktig rolle i å hemme utviklingen av oksidativt stress (Shi et al., 2014; Liu C. et al., 2015). DET ble funnet AT KLF4 kan fremme cellene apoptose indusert AV H2O2, denne handlingen er sannsynligvis forårsaket av økt bax-uttrykk og redusert bcl-2-uttrykk (Li et al., 2010). Quercetin kan redusere KLF4-uttrykk i humane neuroblastom sh-SY5Y-celler, og øke uttrykket for bcl-2 / bax-forhold. Videre Kan Quercetin moderere apoptose frekvensen AV sh-5YSY celle og redusere caspase – 3 enzymaktivitet (Xi et al., 2012). En nylig studie undersøkte den nevrobeskyttende effekten av mitogenaktivert protein (MAP) kinase 5 (ERK5) mot oksidativt stress. Aktivering AV ERK5 kan delvis redusere H2O2-indusert hippocampal neurons død og øke NGF-og PC-indusert neuroprotection (Su et al., 2014). Nils et al. brukt en mutant AV MEK5 (MEK5D)for å studere ERK5-aktivert transkripsjon og funksjonelle responser i humane endotelceller, og identifisert KLF4 var en roman nedstrøms ERK5 mål (Ohnesorge et al ., 2010). Det ble funnet at overuttrykk AV KLF4 kan undertrykke tnf-mediert inflammatorisk respons og redusere leukocyttadhesjon og basalcelleapoptose. Disse resultatene bekrefter AT KLF4 har antiinflammatoriske og anti-apoptotiske egenskaper (Ohnesorge et al., 2010). Påfølgende eksperimenter har vist at forsvinningen av cerebral cavernous misdannelse 1 (CCM1)i endotelceller aktiverer ERK5 via MEKK3-MEK5 signalveien og øker klf4 uttrykk (Cuttano et al., 2016). ERK5 spiller en medierende rolle i preconditioning (PC) og nerve vekstfaktor (NGF)opp-regulert uttrykk FOR KLF4 (Su et al., 2014). I Tillegg kan RNAi-mediert nedslag av KLF4 også redusere NGF – eller PC-indusert nevrobeskyttelse. Overuttrykk AV KLF4 fører til høyere bcl-2 / bax-forhold I H2O2-stressede celler (Su et al., 2014). Over-uttrykt KLF4 akselererer endringer i bcl – 2 og bax ved å kombinere med sin tilsvarende promoter (Li et al., 2010). ERK5 / KLF4 kaskade kan fungere som en pivot i ulike veier som beskytter nevroner fra oksidativt stress-indusert død (Su et al., 2014).
Oksidativt stress har vært ansett å være nært knyttet til mange degenerative sykdommer. KLF4 spiller viktige roller i å opprettholde genomisk stabilitet i oksidativt stress. KLF4 og ERK5 virker sammen for å beskytte nevroner mot oksidativ stress-indusert apoptose. DERFOR KAN KLF4 fungere som et terapeutisk mål for å virke mot oksidativt stress når det aktiveres. Det har blitt rapportert at statinmedisiner kan aktivere ERK5, noe som fører til uttrykk FOR KLF4 og dets avhengige gener (Ohnesorge et al., 2010), men mekanismen forblir uklar, OG KLF4-relaterte oppstrøms-og nedstrøms målgener er mindre studert i oksidativt stress, det er behov for videre studier.
Rolle KLF4 I Axon Regenerering
Tidlig axon tap er et vanlig trekk ved nevrodegenerative sykdommer. Synaptisk tap og transport svekkelse I AD kan forårsake kognitive svekkelser (Holtzman et al.( 2011; Coleman, 2013). Graden av deklarativ minneskade er relatert til synaptisk tetthet i hippocampus og cortex. Oppløselige Aß oligomerer reduserer glutamatopptak og fremmer synaptisk dysfunksjon, forstyrrer synaptisk plastisitet (Li et al., 2009). Derfor er det spesielt viktig å studere hvordan man reparerer axonene i CNS. I retinale ganglionceller har axoner en sterk evne til å vokse og regenerere under tidlig utvikling, men i CNS hos voksne pattedyr mister axoner regenereringskapasiteten og nevronene kan oppgradere til å dø eller atrofi (Goldberg og Barres, 2000; Goldberg et al., 2002).
KLF4 spiller en viktig rolle i å hemme axonvekst. I embryonale RGCs kan OVERUTTRYKK AV KLF4 redusere prosentandelen av neurittforlengelse, lengden på axoner og dendriter og neurittforgrening. Dessuten ble det funnet at overuttrykk AV KLF4 kan redusere langsiktige postnatal axon vekstrater, men klarte ikke å redusere kortsiktige axon vekstrater (Moore et al ., 2009; Jørgen et al., 2014). Senere studier har funnet at axonbuntene AV KLF4-cKO-mus var tykkere enn kontrollmus (Fang et al., 2016). I tillegg kan fjerning AV KLF4-uttrykk under utvikling øke reproduksjonspotensialet til voksne RGCs. I TILLEGG hadde KLF4 som manglet det c-terminale DNA-bindingsdomenet ingen effekt på aksonveksten. Det var ingen innvirkning på overlevelse av celler etter at retinale ganglionceller ble skadet dersom KLF4 ble banket ut (Moore et al., 2009).
KLF4 kan også påvirke aksonal regenerering. En nylig studie rapporterte at reduksjonen AV KLF4-uttrykk i voksne retinale ganglionceller fremmet axonregenerering gjennom JAK-STAT3-bane (Qin et al., 2013). KLF4 økte fosforyleringen AV STAT3, og regulerte aksonveksten via JAK-STAT signalering (Qin Og Zhang, 2012). Under behandling av cytokiner fosforyleres medlemmer av stat-familien av proteiner ved karboksy-terminale tyrosin-og serinsteder i cellen for å danne en stabil dimer. Denne modifikasjonen forbedrer transkripsjon av celleassosierte gener (Yuan et al., 2005). Samspillet MELLOM KLF4 OG STAT3 på cytokinindusert fosforylering av tyrosin705 hemmer ekspresjonen AV STAT3 ved å hemme bindingen AV STAT3 TIL DNA (Qin et al., 2013). KLF4 knockdown forbedrer åpenbart axons regenerering i retinale ganglionceller etter skade på optisk nerve, og forhindrer nerve fra skade etter mild hjerneskade. Handlingene er formidlet av en reduksjon i p-p53 og en økning i pstat3 nivåer. KLF4 regulerer positivt neuronal apoptose via p53-og JAK-STAT3-veiene, OG KLF4 regulerer negativt aksonal reparasjon via JAK-STAT3-banen (Cui et al ., 2017).
derfor antydet vi at i AD kan aksonal regenerering oppnås ved å endre uttrykket FOR KLF4 eller endre intracellulære relaterte signalveier, og kontrollere AD-progresjon ved å redusere manglende aksoner eller redusere aksonal dysfunksjon. Men hvordan DU bruker klf4 transkripsjonsfaktoren i potensiell terapi, trenger fortsatt videre utforskning.
Klf4s Rolle I Jernakkumulering
Jern er mye funnet i biologiske systemer, jernrelaterte metalloproteinaser spiller en nøkkelrolle i transport av oksygen, overføring av elektroner og katalysering av biokjemiske reaksjoner (Aisen et al ., 2001). Imidlertid kan overskudd av jern utover det normale fysiologiske området skade menneskers helse(Adlard Og Bush, 2006). Studier har funnet at jerninnholdet i hippocampus er negativt korrelert med ytelsen til minnetester (Ding et al., 2009). Økt jernbelastning i hjernen akselererer dannelsen Av Enß plakk og hyperfosforylerte tau tangles, samtidig som det øker oksidativt stress (Peters et al., 2015). Jern, som har en høy grad av permeabilitet, fremmer nervevekst og celle-til-celle-forbindelser under hjernens utvikling (Dallman Og Spirito, 1977).
en fersk studie viste at fysiologisk stress forårsaket aktivering AV KLF4-HCP1 signalveien og økt heme opptak (Li H. et al., 2017). Heme står for 95% av det funksjonelle jernet i menneskekroppen. Det er en av hovedkomponentene i heme oxygenase (Hooda et al., 2014; Mikkelsen et al., 2018). Øke aktiviteten av oxygenase-1 kan forsinke oksidasjon av den aldrende hjernen (Verdile et al., 2015; Serini Og Calviello, 2016; Kurucz et al., 2018). Dette har en lettelse effekt PÅ AD. Fysiologisk stress induserer glukokortikoid nivå økning, glukokortikoid øker hemebærer protein 1 (HCP1) uttrykk via KLF4, OG DERETTER HCP1 fremmer heme opptak (Li H. et al., 2017). Glukokortikoid og KLF4 regulerer antiinflammatoriske gener sammen, og celler med lavt glukokortikoidinnhold kan ikke fullt ut indusere KLF4-uttrykk (Sevilla et al ., 2015). KLF4-indusert økning i heme inntak fører til jernakkumulering i hjernen. Jern fremmer utgivelsen AV ROS (Tronel et al., 2013). Jern element forbedrer hjernen oksidativt stress i rotter under psykisk stress (Yu et al., 2011). HCP1 kan derfor reguleres AV KLF4 og glukokortikoid sammen. Økende HCP1 øker hemeopptaket, noe som fører direkte til jernakkumulering i hjernen, forverrer oksidasjon, øker apoptose eller dysfunksjon og forverrer hjerneskade.
det er generelt akseptert at minnet og lærevansker er de viktigste symptomene PÅ AD. Et stort antall kliniske data har vist at Enß plakkbelastning og jernakkumulering respons på utvikling av læring og kognitiv dysfunksjon I AD (van Bergen et al ., 2018). Nylig publiserte data har antydet at høydose jern øker Enß deponering og demper læring og minne hos mus(Guo et al ., 2013). Kliniske studier har vist at jernholdig mikroglia er funnet i hippocampus HOS AD-pasienter under magnetisk resonansavbildning (Zeineh et al., 2015). Mikroglia kjøper jern fra overføring eller ikke-overføring, ekstracellulære og intracellulære kilder (McCarthy et al., 2018). Selektivt OG vedvarende KLF4-uttrykk kan induseres i kjernen og cytoplasma av iskemiske hippocampale reaktive astrocytter (Park et al., 2014). Studier har vist AT KLF4 fungerer som en transkripsjonell repressor. Det nedregulerer uttrykket AV ELG-3, og DERETTER hemmer ELG-3 uttrykket AV HO-1 (Tsoyi et al., 2015). Heme oxygenase-1 (HO-1) er et stressprotein som nedbryter heme til bilirubin, fritt jern og karbonmonoksid. Oppregulering AV HO-1 i astrocytter kan føre til unormal jernavsetning og mitokondriell dysfunksjon i hjernen, noe som fører til redusert kognitiv evne (Schipper, 1999, 2004). DERFOR KAN KLF4 være involvert i prosessen med jernakkumulering i astrocytter, forverrende oksidasjon i AD og forverrende hjerneskade.
Konklusjon
KLF4 er kjent for å spille en sentral rolle i regulering av celleproliferasjon, apoptose og differensiering. Tidligere studier har fokusert på regulering AV KLF4 i flere viktige nevrofysiologiske prosesser, inkludert nevroinflammasjon, nevrobeskyttelse og synaptisk regenerering. NYLIG HAR KLF4 vist seg å spille en viktig rolle i PATOGENESEN AV AD. I denne artikkelen vurderer VI KLF4S rolle i nevrobeskyttelse og nevrogenese I AD.
KLF4 er ikke bare en regulator for regulering av celleproliferasjon og differensiering, men også et potensielt mål for regulering av immunresponser. KLF4 kan regulere negative inflammatoriske faktorer og fremme inflammatorisk respons, og har stor effekt på uttrykket av astrocyt nukleær mikroglia. I TILLEGG KAN KLF4 og ERK5 handle sammen for å utøve nevrobeskyttende tiltak. Videre kan axonregenerering oppnås ved å endre innholdet av spesifikke transkripsjonsfaktorer, intracellulære hemmere eller endre intracellulære signalveier. Slå UT KLF4 kan forbedre axon regenerering og akselerere axon vekst. Reduksjon AV KLF4-uttrykk fremmer aksonregenerering gjennom JAK-STAT3-banen, OG KLF4 fremmer JAK-STAT3-banen for ytterligere aksonregenerering. DERFOR KAN KLF4 være involvert i prosessen med antiinflammatorisk, anti-apoptose, aksonregenerering og jernakkumulering i CNS, som spiller en sentral rolle i AD-generasjonen. Disse funnene tyder på AT KLF4 representerer et potensielt terapeutisk mål for AD. Imidlertid er de dype cellulære og molekylære mekanismene for EFFEKTEN AV KLF4 på AD fortsatt uklare, og det er behov for ytterligere undersøkelser.
Forfatterbidrag
zqc, XHZ og YJ skrev manuskriptet. SHG og JYL endret rammen av manuskriptet. BJL og RJC ga de kritiske revisjonene. Alle forfattere godkjente den endelige versjonen av manuskriptet for innsending.
Finansiering
dette arbeidet ble støttet av tilskudd Fra Natural Science Foundation Of China (NSFC) (81871070, 31571126, 31471120) Og jilin Science And Technology Agency finansiering (Grant Nr.20180519003jh, 20180414051GH, 20170414034GH og 20180414050gh).
Interessekonflikt
forfatterne erklærer at forskningen ble utført i fravær av kommersielle eller økonomiske forhold som kan tolkes som en potensiell interessekonflikt.
Cacabelos, R. (2018). Har det vært forbedringer I Alzheimers sykdom drug discovery de siste 5 årene. Exp. Opin. Narkotika Discov. 13, 523–538. doi: 10.1080/17460441.2018.1457645
PubMed Abstrakt / Fulltekst / Google Scholar
Liu, Y., Shi, J., Og Chen, X. (2015). Identifisering av nye mål for sesongmessig allergisk rhinitt under og utenfor pollensesongen ved mikroarray analyse. Acta Otolaryngol. 135, 1330–1336. doi: 10.3109 / 00016489.2015.1067906
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
Serini, S., Og Calviello, G. (2016). Reduksjon av oksidativt / nitrosativt stress i hjernen og dets involvering i den nevrobeskyttende effekten av n – 3 PUFA I Alzheimers sykdom. Curr. Alzheimers Res. 13, 123-134. doi: 10.2174/1567205012666150921101147
PubMed Abstrakt | CrossRef Fulltekst / Google Scholar