- Introduzione
- Materiali e metodi
- Preparazione di campioni
- Diluizione a gradiente e amplificazione a spostamento multiplo
- Costruzione e sequenziamento della libreria
- Analisi dei dati
- Controllo qualità sequenza e filtraggio
- Assegnazione tassonomica e diversità
- Leggi Assemblaggio, previsione genica e annotazione
- Numeri di adesione della sequenza nucleotidica
- Risultati
- Progettazione sperimentale e sequenziamento
- Annotazione tassonomica
- Assemblaggio metagenomico, previsione genica e annotazione funzionale
- Discussione
- Conclusione
- Contributi dell’autore
- Dichiarazione sul conflitto di interessi
- Riconoscimento
- Materiale supplementare
Introduzione
Koumiss, anche chiamato chige, chigo, arrag, o airag, in lingua mongola, è un tipo di prodotto lattiero-caseario fermentato tradizionale. È stato un alimento popolare in Mongolia e nella Mongolia interna della Cina per secoli (Zhang e Zhang, 2011). Le persone in queste regioni consumavano koumiss durante le grandi feste e le offerte sacrificali (Zhang e Zhang, 2011). La prima testimonianza della produzione di koumiss può essere fatta risalire alla dinastia Han (BC202-AD202). Questo prodotto aveva raggiunto la popolarità diffusa durante la dinastia Yuan (AD1271-AD1368) (Zhang et al., 2010b). Oggigiorno, il koumiss è un alimento comune per la popolazione locale della Mongolia e della Mongolia interna, anche se solo in poche di queste aree, è fabbricato su scala industriale. Koumiss non solo fornisce nutrienti ricchi, tra cui un alto contenuto di aminoacidi essenziali e vitamine, ma si ritiene anche che allevii una vasta gamma di condizioni mediche ed è benefico per la cura postoperatoria (Jagielski, 1877; Thompson, 1879).
Tradizionalmente, il koumiss è comunemente prodotto in botti di legno, contenitori fatti di pelle animale o urne. La fermentazione avviene naturalmente a temperatura ambiente dopo l’aggiunta di latte di cavalla filtrato nel contenitore con il vecchio koumiss, che funge da coltura iniziale (Zhang e Zhang, 2011). Koumiss è una buona fonte di nuovi batteri del potenziale biotecnologico (Zhang et al., 2010a; Pan et al., 2011). Pertanto, è di intenso interesse esplorare e preservare il maggior numero possibile di batteri koumiss associati alla fermentazione. Durante gli ultimi decenni, sono stati eseguiti numerosi studi per indagare la comunità batterica koumiss (Wu et al., 2009; Hao et al., 2010), principalmente studiato con metodi basati su cultura, biologia molecolare e pirosequenziamento (Sun et al., 2010). Tra questi diversi approcci, il metodo basato su pyrosequencing ha fornito il profilo microbiota più completo di koumiss indipendente dai tratti fenotipici e dai problemi di coltivabilità dei singoli microbi. Tuttavia, lo spettro dei geni funzionali codificati dai batteri koumiss e le loro capacità fermentative rimangono scarsamente caratterizzati, in particolare per le rare popolazioni microbiche.
Il presente studio ha utilizzato la tecnica della genomica a cellule singole per analizzare i metagenomi batterici di 10 campioni di koumiss raccolti dalla Mongolia e dalla Mongolia interna della Cina. Il lavoro attuale ha applicato tecnologie all’avanguardia nello studio della diversità batterica nei prodotti lattiero-caseari. Il nostro lavoro ha dimostrato la fattibilità di scoprire taxa a bassa abbondanza applicando l’approccio metagenomico a singola cella. I risultati incoraggianti promuoverebbero lo sviluppo e l’applicazione di nuovi approcci per affrontare i problemi in un campo di ricerca tradizionale.
Materiali e metodi
Preparazione di campioni
Un totale di 10 campioni koumiss sono stati raccolti dalla Mongolia (MG14, MG15, MG16, MG17 e MG18) e dalla Mongolia interna della Cina (NM17, NM18, NM19, NM20 e NM21) per lo studio metagenomica. I campioni sono stati raccolti in modo asettico e sono stati trasportati in ghiaccio secco.
Un millilitro di ciascun campione è stato pretrattato secondo la metodologia descritta in Ward et al. (2013) con alcune modifiche. In breve, i campioni sono stati scongelati in un bagno di ghiaccio per 3-5 min. Dopo che i campioni si sono sciolti, sono stati sottoposti a centrifugazione a bassa velocità per rimuovere impurità e grumi di cellule eucariotiche. Le cellule procariotiche sono state poi pellettate dai sieri del latte mediante centrifugazione a 13.000 × g per 15 min. I pellet sono stati nuovamente sospesi in 2 ml di soluzione salina tampone fosfato (PBS) con 1% Triton X-100 e incubati per 2 ore a 37°C per lisare eventuali cellule eucariotiche rimanenti. Successivamente, i batteri sono stati pellettati mediante centrifugazione a 13.000 × g per 15 min e i pellet sono stati nuovamente sospesi in 500 µL di PBS. Infine, la fase di centrifugazione è stata ripetuta ancora una volta per lavare le cellule batteriche.
Diluizione a gradiente e amplificazione a spostamento multiplo
Per rilevare i batteri a bassa abbondanza, la sospensione batterica derivata da ciascun campione di koumiss è stata diluita in serie per la successiva reazione di amplificazione. Il numero di cellule in ogni campione è stato approssimativamente stimato al microscopio (Nikon, Tokyo, Giappone) utilizzando una camera di conteggio delle cellule (Qiujing, Shanghai, Cina). La fase di diluizione è stata continuata fino a quando il numero di cellule in ciascuna sospensione batterica ha raggiunto circa 100. L’amplificazione a spostamento multiplo delle celle diluite è stata eseguita utilizzando il kit a cella singola REPLI-g (Qiagen, Germantown, MD, USA) secondo le istruzioni del produttore.
Costruzione e sequenziamento della libreria
Il DNA amplificato è stato tagliato in modo casuale e sono stati selezionati i frammenti di circa 500 bp. Dopo la costruzione della biblioteca, PerkinElmer LabChip® GX Touch e StepOnePlusTM Real-Time PCR System sono stati utilizzati per l’ispezione della qualità della biblioteca. Infine, le letture accoppiate a 125 bp sono state sequenziate sulla piattaforma Illumina HiSeq 2500 secondo le istruzioni del produttore.
Analisi dei dati
Controllo qualità sequenza e filtraggio
Le letture raw generate dal sequencer potrebbero contenere letture artificiali di contaminazione dell’adattatore durante la costruzione della libreria. Pertanto, sono stati eseguiti tre passaggi per ottenere un set di dati di lettura pulita di alta qualità: (1) eliminazione delle letture causate dalla contaminazione dell’adattatore; (2) rimozione di letture con un punteggio medio inferiore a un punteggio phred di Q30, che è stato considerato come il taglio più basso per una base di alta qualità; (3) rimozione di letture con un eccesso significativo di “N” (≥5% della lettura). L’analisi a valle era basata sui dati puliti. Inoltre, è stata calcolata la qualità di base statistica, basata su Q30 e il contenuto di GC.
Sono stati effettuati allineamenti contro il genoma ospite per rimuovere le sequenze di contaminanti originate dall’ospite. Qualsiasi lettura originata dall’ospite è stata scartata prima di un ulteriore confronto con sequenze di riferimento del genoma di batteri (o virus). Per ottenere risultati più accurati, il modello MEM di Burrows–Wheeler aligner (BWA) (versione 0.97 a) è stato utilizzato negli allineamenti (Li e Durbin, 2009).
Assegnazione tassonomica e diversità
Il software web Metaphlan è stato utilizzato per l’assegnazione tassonomica ai livelli di genere e specie (Segata et al., 2012). Per confrontare la diversità delle specie all’interno e tra i campioni, abbiamo analizzato la diversità alfa e beta dal pacchetto relativo a R.
Leggi Assemblaggio, previsione genica e annotazione
Per ottenere informazioni più complete, abbiamo assemblato i frammenti tranciati in genoma (contigs). Tuttavia, a causa della presenza di più specie, che è comune nei campioni metagenomici, abbiamo migliorato il metodo di assemblaggio del genoma bioinformatico normalmente utilizzato per l’analisi di singole specie integrando SPAdes (Versione 3.6.2), script interni e database metagenomici (Zerbino e Birney, 2008; Nurk et al., 2013).
Il software MetaGeneMark è stato utilizzato per la previsione genica dei contig assemblati (Noguchi et al., 2006). I geni ridondanti sono stati rimossi usando CD-HIT con la copertura dell’identità 90 e 95% (Li e Godzik, 2006; Fu et al., 2012). Le abbondanze relative dei geni sono state determinate allineando le letture di sequenziamento di alta qualità al catalogo dei geni utilizzando la stessa procedura. L’analisi discrepante a valle era basata sulle abbondanze relative del gene. L’annotazione genica è stata eseguita allineando le sequenze di alta qualità contro diversi database pubblici (ovvero database NCBI non ridondante, NR; Cluster di gruppi ortologhi di proteine, INGRANAGGI; Kyoto Encyclopedia of Genes and Genomes, KEGG) utilizzando BLAST (Altschul et al., 1997). Una ricerca di dominio è stata eseguita utilizzando Interproscan (Mulder e Apweiler, 2008).
Numeri di adesione della sequenza nucleotidica
I dati di sequenza riportati in questo studio sono stati depositati nel database SRA (Accession No.SRP083102).
Risultati
Progettazione sperimentale e sequenziamento
Per rilevare i batteri a bassa abbondanza in koumiss, la tecnica di amplificazione monocellulare è stata utilizzata per analizzare i metagenomi dei campioni. Tre sospensioni batteriche, ciascuna di circa 100 cellule, sono state ricavate da un campione di koumiss indipendente mediante diluizione seriale. Sono state analizzate in totale 30 sospensioni diluite. A ciascun campione diluito è stato assegnato un codice di esempio diverso, ovvero il numero identificativo del campione seguito da 1, 2 o 3, che rappresenta le tre diluizioni separate. Sulla base di una premessa che alcune specie rare saranno presenti in una delle diluizioni, è stata effettuata un’amplificazione a spostamento multiplo delle cellule; e sono stati generati circa 5 Gb di dati per ogni sospensione batterica koumiss.
Un totale di 1.040.323.864 letture grezze sono state generate dai 10 campioni di koumiss (per un totale di 30 sospensioni batteriche). Il numero medio di letture per ogni sospensione a 100 celle è stato di 34.677.462 (Tabella supplementare S1). Dopo il taglio e il filtraggio delle sequenze non qualificate, abbiamo ottenuto 1.018.381.702 letture pulite per tutti i campioni (tabella supplementare S1). I valori di Shannon index, Simpson index, Chao1 index e il numero di specie osservate (Figure 1-4) hanno mostrato che la maggior parte dei campioni di koumiss aveva un’alta biodiversità batterica. Le curve di diversità di Shannon-Wiener hanno mostrato che la profondità di sequenza era adeguata per tutti i campioni (Figura 1).
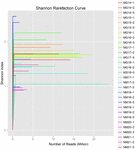
FIGURA 1. Curve di rarefazione di Shannon che stimano la diversità microbica dei campioni di koumiss.
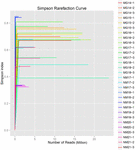
FIGURA 2. Curve di rarefazione di Simpson che stimano la diversità microbica dei campioni di koumiss.
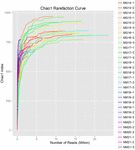
FIGURA 3. Curve di rarefazione Chao1 che stimano la diversità microbica dei campioni di koumiss.
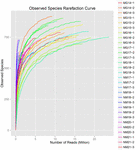
FIGURA 4. Indice di rarefazione delle specie osservato che stima la diversità microbica dei campioni di koumiss.
Annotazione tassonomica
Le sequenze di alta qualità sono state assegnate a diversi livelli tassonomici per consentire un’analisi approfondita delle comunità batteriche del campione. Con riferimento ad alcuni studi pubblicati sulla biodiversità di koumiss (Wu et al., 2009; Hao et al., 2010; Sun et al., 2010), abbiamo classificato i batteri associati koumiss noti e precedentemente non segnalati come taxa comuni e rari, rispettivamente.
Le sequenze di alta qualità rappresentavano 13 generi diversi (Figura 5). Tre di loro avevano un’abbondanza relativa media di oltre l ‘ 1%, tra cui Lactobacillus (L.), Lactococcus e Streptococcus. In particolare, Lactobacillus e Lactococcus erano i due generi più abbondanti trovati nel koumiss. La percentuale di Lactobacillus nei campioni variava dal 52,72 al 99,96%. Due membri di questo genere, L. helveticus e L. kefiranofaciens, erano dominati tra la maggior parte dei campioni di koumiss (Figura 6). La specie L. buchneri era presente principalmente nei campioni mongoli, mentre Lactococcus lactis è stato rilevato nella maggior parte dei campioni indipendentemente dalla regione di campionamento.
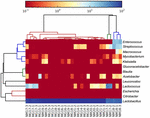
FIGURA 5. Mappa termica che mostra l’abbondanza relativa di batteri rilevati nei campioni di koumiss a livello di genere.
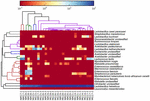
FIGURA 6. Mappa termica che mostra l’abbondanza relativa di batteri rilevati nei campioni di koumiss a livello di specie.
Assemblaggio metagenomico, previsione genica e annotazione funzionale
L’assemblaggio delle letture ha portato a una lunghezza di assemblaggio di 614,392,623 bp. I valori N50 per le assemblee variavano da 5.596 a 35.200 bp (Tabella supplementare S2). Il numero di geni previsti nei campioni di koumiss variava da 10.347 a 34.547, con una lunghezza media compresa tra 647,82 e 985,33 bp (Tabella supplementare S3). Sebbene le analisi funzionali empiriche dei geni fossero al di là dello scopo dello studio attuale, abbiamo annotato i microbiomi batterici di koumiss utilizzando i database COG e KEGG, che prevedevano la funzione genica in gran parte basata sull’omologia delle sequenze.
Un totale di 545 corrispondenze nell’output di annotazione ha mostrato un’elevata omologia ai geni di utilizzo del lattosio (categoria INGRANAGGI di carboidrati e metabolismo, G), e alcuni di essi si trovavano all’interno dello stesso contig (Tabella 1).
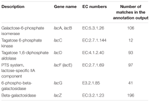
TABELLA 1. Geni correlati al metabolismo del lattosio annotati nel metagenoma batterico koumiss.
Alcune altre sequenze potrebbero codice per putativi geni all’interno di COGs categoria di aminoacidi trasporto e metabolismo (E), comprese le sequenze che corrispondono alla caseina degradante proteinases, l’Opp di sistema per l’assunzione di oligopeptidi di 4-18 residui, e aminopeptidasi (ad esempio, leucyl aminopeptidasi, peptidilica-dipeptidasi Una, aminopeptidasi N, prolina iminopeptidase, e endopeptidasi). Sebbene alcune sequenze condividessero elevate somiglianze con le aminotransferasi specifiche per arginina, aspartato, metionina e isoleucina, è stata identificata solo una corrispondenza significativa, che corrispondeva a un presunto dominio di classe I/classe II di S. macedonicus (IPR004839) contenente aminotransferasi specifica per tirosina e fenilalanina. Infine, un certo numero di sequenze condivise alta omologia alle liasi aminoacidiche, tra cui S-ribosilomocisteina liasi, argininosuccinato liasi, aspartato ammoniaca-liasi, cistationina gamma-liasi, istidina ammoniaca-liasi, e O-acetilomoserina (tiolo)-liasi.
Discussione
Koumiss è un popolare prodotto lattiero-caseario tradizionale fermentato in Mongolia e Mongolia interna della Cina. Sebbene siano stati condotti numerosi studi per indagare la diversità batterica in koumiss, sono state ottenute poche informazioni sulla capacità genetica del microbiota di koumiss. Qui, abbiamo applicato la tecnica di amplificazione monocellulare per analizzare i metagenomi batterici koumiss, concentrandosi in particolare sulla popolazione batterica a bassa abbondanza.
L’approccio metagenomico tipico è stato precedentemente applicato per descrivere il microbiota koumiss. Tuttavia, a causa dell’elevato costo per ottenere una sequenza profonda, la rara popolazione di microbiota nei campioni è spesso coperta in modo inadeguato. Pertanto, entrambi i metagenomi filogenici e funzionali dei batteri di minoranza rimangono limitati. Il nostro metodo di amplificazione a cella singola prevedeva la diluizione seriale di campioni in sospensioni a 100 celle. A causa del basso numero di cellule presenti nei campioni di koumiss diluiti, solo una quantità limitata di DNA sarebbe stata estratta. La fase di amplificazione ha aumentato la quantità di materiali di DNA da analizzare e quindi ha facilitato l’analisi del metagenoma di campioni contenenti quantità minime di DNA. Uno svantaggio di questo metodo era la difficoltà nel garantire che le sequenze di ciascun taxon fossero ugualmente amplificate nel processo; pertanto, i risultati ottenuti qui potrebbero riflettere solo le abbondanze relative delle sequenze ma non le quantità assolute di taxa identificati o geni funzionali. Tuttavia, questo avrebbe scarso effetto sulla nostra analisi poiché il presente studio differisce da altri lavori pubblicati nel focalizzare la popolazione batterica rara. I dati generati da questo lavoro forniscono informazioni complementari alla popolazione sottorappresentata. Riteniamo che l’approccio attuale sia adatto per l’analisi futura della diversità batterica per diversi tipi di ambienti ecologici.
Il microbiota batterico koumiss è costituito principalmente da batteri lattici (LABORATORIO) e alcuni batteri dell’acido acetico (Zhang e Zhang, 2011). Come previsto, il nostro set di dati conteneva principalmente sequenze che rappresentano il LABORATORIO e i batteri dell’acido acetico. Sequenze corrispondenti a Lactobacillus helveticus dominavano su tutti i campioni. Inoltre, sequenze che rappresentano la specie, L. kefiranofaciens. L. buchneri. L. kefiranofaciens. Enterococcus (E.) casseliflavus. E. fecalis. E. fecio. Leuconostoc mesenteroides. Sono stati trovati anche Lactococcus lactis e Acetobacter pasteurianus. L’identificazione di sequenze di diversi taxa potrebbe non essere sufficiente per mostrare la vitalità dei batteri, tuttavia riflette la comunità batterica ad un certo punto del processo di fermentazione. Miyamoto et al. (2010) suggerisce che la prevalenza batterica nei prodotti fermentati finali è correlata alla loro tolleranza allo stress acido. Generalmente, i lattobacilli hanno una tolleranza acida più elevata rispetto ai lattococchi, il che può spiegare l’elevata abbondanza relativa di sequenze di lattobacilli presenti nel nostro set di dati. D’altra parte, il frequente verificarsi di L. helveticus in koumiss coincise con l’osservazione della dominanza delle sequenze di L. helveticus.
Nel nostro set di dati, alcune sequenze corrispondevano a L. otakiensis, che è una specie rara che non è mai stata segnalata in koumiss o in altre nicchie da latte. La specie è stata inizialmente descritta e isolata dalla soluzione di decapaggio non salata utilizzata nella produzione di sunki, un sottaceto tradizionale giapponese (Watanabe et al., 2009). È stato scoperto dal profilo di polimorfismo della lunghezza del frammento amplificato basato sul gene recA. Da allora, questa specie non è stata segnalata per essere associata ad altre nicchie legate al cibo. Quindi, è probabile che questo batterio appartenga alla flora autoctona dei sottaceti. Tuttavia, non possiamo escludere la possibilità che non sia stato trovato semplicemente a causa della sensibilità del metodo di rilevamento utilizzato. Lactobacillus otakiensis può produrre aminoacidi a catena d-ramificata, come d-leucina, d-allo-isoleucina e d-valina (Doi et al., 2013). Ha il potenziale per essere utilizzato nel miglioramento delle caratteristiche di produzione di alcuni alimenti fermentati (Kato et al., 2011).
Il nostro studio ha anche identificato sequenze che rappresentano la specie S. macedonicus, che non è mai stata segnalata come batterio associato a koumiss. Invece, è una cultura starter presente nei formaggi ovini e caprini greci (Georgalaki et al., 2000). Alcuni membri di questa specie sono in grado di produrre esopolisaccaridi, batteriocine (Vincent et al., 2001; Anastasiou et al., 2015) e acido gamma-aminobutirrico (Franciosi et al., 2015). Anche se questa specie è spesso isolata da alimenti fermentati, la nicchia originale di S. macedonicus è stata controversa (Guarcello et al., 2016). Non fino a poco tempo fa, Papadimitriou et al. (2015) identificato un plasmide acquisito, pSMA198, da Lactococcus lactis. Il plasmide è stato probabilmente trasferito attraverso un evento di scambio genetico ancestrale all’interno di un ambiente di prodotti lattiero-caseari, suggerendo l’origine casearia di S. macedonicus (Papadimitriou et al., 2015). Simile a S. thermophilus. S. macedonicus è strettamente correlato ai commensali e agli agenti patogeni opportunistici del complesso S. bovis/S. equinus.
L’identificazione di sequenze che rappresentano le coppie di ruminococco della specie è stata inaspettata, in quanto questo batterio si trova solitamente nell’ambiente intestinale. È un normale microbo intestinale umano che può degradare gli oligosaccaridi della mucina mediante produzione costitutiva di glicosidasi secretorie (Hoskins et al., 1985). Recenti prove cliniche mostrano che l’abbondanza fecale di questa specie è alterata nei bambini con disturbo dello spettro autistico; tuttavia, il suo ruolo nel disturbo rimane poco chiaro (Wang et al., 2013).
Sorprendentemente, alcune delle sequenze rappresentavano potenziali agenti patogeni. Ad esempio, la polmonite da Klebsiella è un patogeno umano opportunistico che risiede in circa il 40% delle viscere umane e animali. Mycobacterium orygis, precedentemente chiamato oryx bacillus, è un membro del complesso Mycobacterium tuberculosis che può causare la tubercolosi umana (Dawson et al., 2012). Queste due specie sono state segnalate nel latte crudo ma non nel koumiss. Pertanto, la loro presenza potrebbe ascrivere alla contaminazione durante la produzione di koumiss convenzionale, specialmente in condizioni di manipolazione non o poco asettiche.
Il metabolismo batterico svolge un ruolo importante nella formazione delle caratteristiche e della qualità del koumiss. Processi microbici come la lipolisi e la proteolisi sono necessari per sintetizzare composti aromatici e aromatici di koumiss (Gesudu et al., 2016). Coerentemente, il metagenoma batterico conteneva sequenze che potenzialmente codificano per la degradazione del lattosio e sistemi proteolitici. A differenza dei percorsi catabolici relativamente semplici del lattosio, i sistemi proteolitici di LABORATORIO sono costituiti da una vasta gamma di enzimi (Chen et al., 2014). Rispetto ad altri koumiss LAB rilevati, la specie dominante L. helveticus è caratterizzata da un’elevata attività proteolitica (Zhang et al., 2015). La maggior parte dei LABORATORI possiede solo una proteinasi dell’involucro cellulare che avvia l’idrolisi della caseina del latte, mentre L. helveticus contiene almeno due di questi enzimi, vale a dire PrtH e PrtH2 (Zhao et al., 2011). Pertanto, le alte proporzioni di sequenze corrispondenti alle forti specie proteolitiche di L. helveticus e ai geni correlati alla proteolisi possono collegarsi al contenuto relativamente elevato di peptidi e amminoacidi liberi in koumiss.
Una difficoltà nella produzione di koumiss su scala industriale è il controllo della percezione del sapore, poiché il koumiss è stato tradizionalmente prodotto dalla fermentazione naturale. Pertanto, è stato difficile definire il sapore e il profilo dei componenti chiave del sapore, in particolare in presenza dei contaminanti naturali. La produzione di componenti chiave del sapore è il risultato della degradazione fermentativa ed enzimatica degli amminoacidi, come gli amminoacidi a catena ramificata, la metionina e gli amminoacidi aromatici (Ardo, 2006). Esempi di componenti aromatici includono aldeidi, acidi organici ed esteri, che sono formati dalla via transaminasi (AT) (Helinck et al., 2004). Questa via è iniziata da una transaminasi che catalizza la conversione di un amminoacido al suo corrispondente acido α-cheto (Helinck et al., 2004). Alcune sequenze nel nostro set di dati corrispondevano a presunte amminoacido aminotransferasi a catena ramificata e aromatica. In particolare, abbiamo trovato un amminoacido aromatico putativo aminotransferasi I nel contig della specie associata a koumiss, S. macedonicus. Poiché il nostro lavoro attuale ha annotato solo il microbioma in silico, i nostri dati non sono sufficienti per dimostrare che queste sequenze genetiche identificate erano effettivamente funzionali per produrre i suddetti composti aromatici koumiss. La presenza di questi geni suggerisce tuttavia che sono alcuni possibili candidati per tali attività fermentative; tuttavia, sarebbe necessario un ulteriore lavoro sperimentale per chiarire i loro esatti ruoli funzionali.
Inoltre, abbiamo trovato sequenze corrispondenti ad altri percorsi di conversione degli aminoacidi tra cui le liasi degli aminoacidi e la treonina aldolasi. L’enzima precedente catalizza la conversione della metionina in metanetiolo (Irmler et al., 2008), con conseguente formazione di dimetil disolfuro e dimetil trisolfuro (Fernandez et al., 2000), mentre quest’ultimo catalizza la conversione della treonina in glicina e acetaldeide (Ott et al., 2000). Allo stesso modo, la semplice localizzazione di queste sequenze all’interno del microbioma di koumiss non può servire come prova definitiva nei loro effettivi ruoli biologici; sarà necessaria una conferma sperimentale futura.
Conclusione
Il presente studio ha utilizzato un metodo metagenomico modificato per analizzare il microbioma batterico dei campioni di koumiss raccolti dalla Mongolia e dalla Mongolia interna della Cina. Abbiamo caratterizzato sia i metagenomi filogenici e funzionali delle specie rare in koumiss; e il nostro set di dati riflette tratti che sono di interesse biotecnologico e potenziale. Il nostro studio ha dimostrato per la prima volta la fattibilità di incorporare le tecniche di amplificazione monocellulare nella rilevazione del microbiota batterico koumiss e dei contaminanti microbici. Le tecniche qui sviluppate possono essere utilizzate in studi futuri per monitorare i cambiamenti nel microbioma koumiss lungo il processo di fermentazione, con particolare attenzione alla popolazione microbica di minoranza. Inoltre, altre tecniche omiche, come la trascrittomica, la metabolomica, possono essere accoppiate all’attuale analisi metagenomica per confermare le funzioni e la capacità metabolica del microbioma koumiss.
Tecnicamente, il passo successivo di questo lavoro sarebbe quello di ottimizzare il metodo corrente. Ad esempio, aumentando la diluizione del campione prima dell’amplificazione del DNA, la possibilità di scoprire batteri rari e nuovi può essere migliorata. D’altra parte, una tecnica di sequenziamento alternativa in grado di generare letture lunghe, come Pacific Biosciences singola molecola, tecnologia di sequenziamento in tempo reale, può essere impiegato per migliorare il processo di assemblaggio del genoma.
Contributi dell’autore
WZ e HZ hanno progettato lo studio. WZ, GY, JY e L-YK hanno scritto il manoscritto. QH, WH, WL, BM e TS hanno eseguito esperimenti. WZ e QH hanno analizzato i dati. Tutti gli autori hanno esaminato il manoscritto.
Dichiarazione sul conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di rapporti commerciali o finanziari che potrebbero essere interpretati come un potenziale conflitto di interessi.
Riconoscimento
Questa ricerca è stata sostenuta dalla National Natural Science Foundation of China (Grant No. 31571815).
Materiale supplementare
Il materiale supplementare per questo articolo può essere trovato online all’indirizzo: https://www.frontiersin.org/article/10.3389/fmicb.2017.00165/full#supplementary-material
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST e PSI-BLAST: una nuova generazione di programmi di ricerca di database di proteine. Acidi nucleici Res. 25, 3389-3402. doi: 10.1093/nar / 25.17.3389
PubMed Abstract / CrossRef Full Text / Google Scholar
Anastasiou, R., Driessche, G. V., Boutou, E., Kazou, M., Alexandraki, V., Vorgias, E. V., et al. (2015). Ceppi ingegnerizzati di Streptococcus macedonicus verso un fenotipo osmotico resistente allo stress mantengono la loro capacità di produrre la batteriocina macedocina in condizioni iperosmotiche. J. Biotechnol. 212, 125–133. doi: 10.1016 / j. jbiotec.2015.08.018
PubMed Abstract | CrossRef Testo completo / Google Scholar
Ardo, Y. (2006). Formazione del sapore per catabolismo aminoacidico. Biotecnologia. Adv. 24, 238-242. doi: 10.1016 / j.biotechadv.2005.11.005
PubMed Abstract | CrossRef Full Text / Google Scholar
Chen, YF, Zhao, WJ, Wu, RN, Sun, ZH, Zhang, WY, Wang, JC, et al. (2014). Analisi del proteoma del Lactobacillus helveticus H9 durante la crescita nel latte scremato. J. Dairy Sci. 97, 7413–7425. doi: 10.3168 / jds.2014-8520
PubMed Abstract | CrossRef Testo completo / Google Scholar
Nel 2012 è stato pubblicato il primo album in studio della band, “The World”, pubblicato nel 2012. Trasmissione di Mycobacterium orygis (M. tuberculosis complex species) da un paziente affetto da tubercolosi a una mucca da latte in Nuova Zelanda. J. Clin. Microbiolo. 50, 3136–3138. doi: 10.1128 / JCM.01652-12
PubMed Abstract | CrossRef Testo completo / Google Scholar
Doi, K., Mori, K., Mutaguchi, Y., Tashiro, K., Fujino, Y., Ohmori, T., et al. (2013). Progetto di sequenza genomica del produttore di aminoacidi a catena D-ramificata Lactobacillus otakiensis JCM 15040T, isolato da un sottaceto tradizionale giapponese. Genoma Announc. 1, e546–e513. doi: 10.1128 / genomeA.00546-13
PubMed Abstract | CrossRef Testo completo / Google Scholar
Fernandez, M., van Doesburg, W., Rutten, GA, Marugg, JD, Alting, AC, van Kranenburg, R., et al. (2000). Analisi molecolari e funzionali del gene metC di Lactococcus lactis, codificante la cistationina beta-liasi. Appl. Ambiente. Microbiolo. 66, 42–48. doi: 10.1128 / AEM.66.1.42-48.2000
PubMed Abstract / CrossRef Full Text / Google Scholar
Franciosi, E., Carafa, I., Nardin, T., Schiavon, S., Poznanski, E., Cavazza, A., et al. (2015). Biodiversità e produzione di acido gamma-amminobutirrico da batteri lattici isolati dai tradizionali formaggi di latte vaccino crudo alpino. Biomed Res. Int. 2015, 625740. doi: 10.1155/2015/625740
PubMed Abstract / CrossRef Testo completo / Google Scholar
Fu, L., Niu, B., Zhu, Z., Wu, S., e Li, W. (2012). CD-HIT: accelerato per il clustering dei dati di sequenziamento di nuova generazione. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
PubMed Abstract | CrossRef Full Text | Google Scholar
Georgalaki, M. D., Sarantinopoulos, P., Ferreira, E. S., De Vuyst, L., Kalantzopoulos, G., and Tsakalidou, E. (2000). Biochemical properties of Streptococcus macedonicus strains isolated from Greek Kasseri cheese. J. Appl. Microbiol. 88, 817–825. doi: 10.1046/j.1365-2672.2000.01055.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Gesudu, Q. M., Zheng, Y., Xi, X. X., Hou, Q. C., Xie, H. Y., Huang, WQ, et al. (2016). Indagare la struttura e la dinamica della popolazione batterica nel koumiss tradizionale della Mongolia interna utilizzando il sequenziamento in tempo reale di una singola molecola. J. Dairy Sci. 99, 7852–7863. doi: 10.3168 / jds.2016-11167
PubMed Abstract | CrossRef Testo completo / Google Scholar
Guarcello, R., Carpino, S., Gaglio, R., Pino, A., Rapisarda, T., Caggia, C., et al. (2016). Una grande applicazione in fabbrica di batteri lattici autoctoni selezionati per la produzione di formaggio Pecorino Siciliano DOP. Microbiolo alimentare. 59, 66–75. doi: 10.1016 / j. fm. 2016.05.011
PubMed Abstract | CrossRef Testo completo / Google Scholar
Hao, Y., Zhao, L., Zhang, H., Zhai, Z., Huang, Y., Liu, X., et al. (2010). Identificazione della biodiversità batterica in koumiss mediante elettroforesi su gel a gradiente denaturante e reazione a catena della polimerasi specie-specifica. J. Dairy Sci. 93, 1926–1933. doi: 10.3168 / jds.2009-2822
PubMed Abstract | CrossRef Testo completo / Google Scholar
Nel 2004 è stato pubblicato il primo album in studio della band. Capacità dei batteri lattici termofili di produrre composti aromatici da aminoacidi. Appl. Ambiente. Microbiolo. 70, 3855–3861. doi: 10.1128 / AEM.70.7.3855-3861.2004
PubMed Abstract | CrossRef Full Text / Google Scholar
Hoskins, L. C., Agustines, M., McKee, W. B., Boulding, E. T., Kriaris, M., e Niedermeyer, G. (1985). Degradazione della mucina negli ecosistemi del colon umano. Isolamento e proprietà dei ceppi fecali che degradano gli antigeni del gruppo sanguigno ABH e gli oligosaccaridi dalle glicoproteine della mucina. J. Clin. Investire. 75, 944–953. doi: 10.1172 | JCI111795
PubMed Abstract / CrossRef Testo completo / Google Scholar
Nel 2008 è stato pubblicato il primo album in studio della band. Clonazione e caratterizzazione di due geni di Lactobacillus casei codificanti una cistationina liasi. Appl. Ambiente. Microbiolo. 74, 99–106. doi: 10.1128 / AEM.00745-07
PubMed Abstract | CrossRef Testo completo / Google Scholar
Jagielski, V. (1877). Il valore di koumiss nel trattamento di nausea, vomito e incapacità di trattenere altri alimenti sullo stomaco. Fr. Med. J. 2, 919-921. doi: 10.1136 / bmj.2.887.919
PubMed Abstract | CrossRef Testo completo / Google Scholar
Kato, S., Ishihara, T., Hemmi, H., Kobayashi, H., e Yoshimura, T. (2011). Alterazioni delle concentrazioni di D-amminoacido e delle strutture di comunità microbiche durante la fermentazione dei vini rossi e bianchi. J. Biosci. Bioeng. 111, 104–108. doi: 10.1016 / j.jbiosc.2010.08.019
PubMed Abstract | CrossRef Testo completo / Google Scholar
Li, H., e Durbin, R. (2009). Veloce e preciso allineamento breve lettura con Burrows-Wheeler transform. Bioinformatica 25, 1754-1760. doi: 10.1093 / bioinformatica/btp324
PubMed Abstract / CrossRef Full Text / Google Scholar
Li, W., e Godzik, A. (2006). Cd-hit: un programma veloce per il clustering e il confronto di grandi insiemi di proteine o sequenze nucleotidiche. Bioinformatica 22, 1658-1659. doi: 10.1093 / bioinformatica / btl158
PubMed Abstract / CrossRef Full Text / Google Scholar
Miyamoto, M., Seto, T., Nakajima, H., Burenjargal, S.,Gombojav, A., Demberei, S., et al. (2010). Elettroforesi su gel a gradiente denaturante analisi di batteri lattici e lieviti nel latte fermentato tradizionale mongolo. Cibo Sci. Tecnologia. Res. 16, 319-326. doi: 10.3136 / fstr.16.319
CrossRef Testo completo / Google Scholar
Nel 2008 si trasferisce a New York. Il database InterPro e gli strumenti per l’analisi del dominio proteico. Curr. Protocollo. Bioinformatica Capitolo 2, Unità2.7. doi: 10.1002 | 0471250953.bi0207s21
PubMed Abstract / CrossRef Full Text / Google Scholar
Noguchi, H., Park, J., e Takagi, T. (2006). MetaGene: ricerca del gene procariotico da sequenze di shotgun del genoma ambientale. Acidi nucleici Res. 34, 5623-5630. doi: 10.1093 | nar / gkl723
PubMed Abstract / CrossRef Full Text / Google Scholar
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A. A., Korobeynikov, A., Lapidus, A., et al. (2013). Assemblaggio di genomi unicellulari e mini-metagenomi da prodotti chimerici MDA. J. Calcolo. Biol. 20, 714–737. doi: 10.1089 / cmb.2013.0084
PubMed Abstract | CrossRef Testo completo / Google Scholar
Ott, A., Germond, J. E., e Chaintreau, A. (2000). Origine dell’acetaldeide durante la fermentazione del latte utilizzando (13) precursori marcati con C. J. Agric. Chimica alimentare. 48, 1512–1517. doi: 10.1021 | jf9904867
PubMed Abstract / CrossRef Full Text / Google Scholar
Nel 2011 è stato pubblicato il primo album in studio. Caratterizzazione di Lactobacillus fermentum SM-7 isolato da koumiss, un potenziale batterio probiotico con effetti ipocolesterolemizzanti. J. Sic. Cibo. Agric. 91, 512–518. doi: 10.1002 / jsfa.4214
PubMed Abstract | CrossRef Testo completo / Google Scholar
Papadimitriou, K., Anastasiou, R., Maistrou, E., Plakas, T., Papandreou, NC, Hamodrakas, SJ, et al. (2015). Acquisizione attraverso trasferimento genico orizzontale del plasmide pSMA198 da Streptococcus macedonicus ACA-DC 198 punti verso l’origine casearia della specie. PLoS UNO 10:e0116337. doi: 10.1371 / giornale.pone.0116337
PubMed Abstract | CrossRef Testo completo / Google Scholar
Nel 2012 è stato pubblicato il primo album in studio della band, “The World”, pubblicato nel 2012. Profilazione della comunità microbica metagenomica utilizzando geni marker specifici del clade. NAT. Metodi 9, 811-814. doi: 10.1038 / nmeth.2066
PubMed Abstract | CrossRef Testo completo / Google Scholar
Sun, Z., Liu, W., Zhang, J., Yu, J., Zhang, W., Cai, C., et al. (2010). Identificazione e caratterizzazione dei lattobacilli dominanti isolati da koumiss in Cina. J. Gen. Appl. Microbiolo. 56, 257–265. doi: 10.2323 / jgam.56.257
PubMed Abstract | CrossRef Testo completo / Google Scholar
Thompson, J. (1879). Il valore di koumiss nello spreco di malattie. Fr. Med. J. 1, 270. doi: 10.1136 / bmj.1.1466.270-c
PubMed Abstract / CrossRef Testo completo / Google Scholar
Nel 2001 è stato pubblicato il primo album in studio della band, “The World”, pubblicato nel 2001. Struttura e proprietà dell’esopolisaccaride prodotto da Streptococcus macedonicus Sc136. Glicobiologia 11, 131-139. doi: 10.1093/glycob | 11.2.131
PubMed Abstract / CrossRef Full Text / Google Scholar
Nel 2013 è stato pubblicato il primo album in studio del gruppo musicale britannico The M. A., pubblicato nel 2013. Maggiore abbondanza di Sutterella spp. e coppie di ruminococco nelle feci di bambini con disturbo dello spettro autistico. Mol. Autismo 4, 42. doi: 10.1186/2040-2392-4-42
PubMed Abstract / CrossRef Testo completo / Google Scholar
Ward, TL, Hosid, S., Ioshikhes, I., e Altosaar, I. (2013). Metagenoma del latte umano: un’analisi della capacità funzionale. BMC Microbiolo. 13:116. doi: 10.1186/1471-2180-13-116
PubMed Astratto | CrossRef Testo Completo | Google Scholar
Watanabe, K., Fujimoto, J., Tomii, Y., Sasamoto, M., Makino, H., Kudo, Y., et al. (2009). Lactobacillus kisonensis sp. ov., Lactobacillus otakiensis sp. ov., Lactobacillus rapi sp. ov. e Lactobacillus sunkii sp. ov., specie eterofermentative isolate da sunki, un sottaceto tradizionale giapponese. Int. J. Syst. Evol. Microbiolo. 59 (Pt 4), 754-760. doi: 10.1099 / ijs.0.004689-0
PubMed Abstract | CrossRef Testo completo / Google Scholar
Wu, R., Wang, L., Wang, J., Li, H., Menghe, B., Wu, J., et al. (2009). Isolamento e selezione probiotica preliminare di lattobacilli da koumiss nella Mongolia interna. J. Microbiolo di base. 49, 318–326. doi: 10.1002 / jobm.200800047
PubMed Abstract | CrossRef Testo completo / Google Scholar
Zerbino, DR, e Birney, E. (2008). Velvet: algoritmi per l’assemblaggio di lettura breve de novo utilizzando grafici de Bruijn. Genoma Res. 18, 821-829. doi: 10.1101 / gr.074492.107
PubMed Abstract | CrossRef Testo completo / Google Scholar
Zhang, W., Sun, Z., Sun, T., e Zhang, H. (2010a). Screening PCR e analisi di sequenza di cluster iol in ceppi di Lactobacillus casei isolati da koumiss. Folia Microbiol. (Praha) 55, 603-606. doi: 10.1007 / s12223-010-0097-3
PubMed Abstract / CrossRef Testo completo / Google Scholar
Zhang, W., Yu, D., Sun, Z., Wu,R., Chen, X., Chen, W., et al. (2010b). Sequenza completa del genoma di Lactobacillus casei Zhang, un nuovo ceppo probiotico isolato dal koumiss tradizionale fatto in casa nella Mongolia interna, in Cina. J. Batteriolo. 192, 5268–5269. doi: 10.1128 / JB.00802-10
PubMed Abstract | CrossRef Testo completo / Google Scholar
Zhang, W., e Zhang, H. (2011). “Fermentazione e koumiss”, in Handbook of Food and Beverage Fermentation Technology, 2nd Edn, ed. Y. H. Hui (Boca Raton, FL: CRC Press), 165-172.
Google Scholar
Zhang, W. Y., Chen, Y. F., Zhao, W. J., Kwok, L. Y., e Zhang, H. P. (2015). Espressione genica del sistema proteolitico del Lactobacillus helveticus H9 durante la fermentazione del latte. Ann. Microbiolo. 65, 1171–1175. doi: 10.3168 / jds.2014-8520
PubMed Abstract | CrossRef Testo completo / Google Scholar
Zhao, W., Chen, Y., Sun, Z., Wang, J., Zhou, Z., Sun, T., et al. (2011). Sequenza completa del genoma del Lactobacillus helveticus H10. J. Batteriolo. 193, 2666–2667. doi: 10.1128 / JB.00166-11
PubMed Abstract / CrossRef Full Text / Google Scholar