- Introducción
- Materiales y métodos
- Preparación de muestras
- Dilución de gradiente y Amplificación de Desplazamiento múltiple
- Construcción y Secuenciación de bibliotecas
- Análisis de datos
- Comprobación de calidad de secuencia y Filtrado
- Asignación taxonómica y Diversidad
- Ensamblaje leído, Predicción de genes y Anotación
- Números de adhesión de Secuencias de nucleótidos
- Resultados
- Diseño experimental y secuenciación
- Anotación taxonómica
- Ensamblaje Metagenómico, Predicción Génica y Anotación Funcional
- Discusión
- Conclusión
- Contribuciones de los autores
- Declaración de Conflicto de Intereses
- Reconocimiento
- Material suplementario
Introducción
El Koumiss, también llamado chige, chigo, arrag o airag, en el idioma mongol, es un tipo de producto lácteo fermentado tradicional. Ha sido un alimento popular en Mongolia y Mongolia Interior de China durante siglos (Zhang y Zhang, 2011). La gente de estas regiones solía consumir koumiss durante las grandes festividades y ofrendas de sacrificio (Zhang y Zhang, 2011). El registro más antiguo de la producción de koumiss se remonta a la dinastía Han (AC202-AD202). Este producto había alcanzado una gran popularidad durante la dinastía Yuan (1271-1368 d.C.) (Zhang et al., 2010b). Hoy en día, el koumiss es un alimento común para la población local de Mongolia y Mongolia Interior, aunque solo en algunas de estas áreas, se fabrica a escala industrial. El koumiss no solo proporciona nutrientes ricos, incluidos altos contenidos de aminoácidos esenciales y vitaminas, sino que también se cree que alivia una amplia gama de afecciones médicas y es beneficioso para el cuidado postoperatorio (Jagielski, 1877; Thompson, 1879).
Tradicionalmente, el koumiss se produce comúnmente en barriles de madera, recipientes hechos de piel de animal o urnas. La fermentación se produce de forma natural a temperatura ambiente después de la adición de leche de yegua filtrada en el recipiente con koumiss antiguo, que sirve como cultivo iniciador (Zhang y Zhang, 2011). El koumiss es una buena fuente de nuevas bacterias con potencial biotecnológico (Zhang et al., 2010a; Pan et al., 2011). Por lo tanto, es de gran interés explorar y preservar la mayor cantidad posible de bacterias koumiss asociadas a la fermentación. Durante las últimas décadas, se realizaron varios estudios para investigar la comunidad bacteriana koumiss(Wu et al., 2009; Hao et al., 2010), estudiada principalmente por métodos basados en cultivo, biología molecular y pirosecuenciación (Sun et al., 2010). Entre estos diferentes enfoques, el método basado en pirosecuenciación ha proporcionado el perfil de microbiota más completo del koumiss independiente de los rasgos fenotípicos y los problemas de cultivabilidad de los microbios individuales. Sin embargo, el espectro de genes funcionales codificados por la bacteria koumiss y sus capacidades fermentativas permanecen mal caracterizados, particularmente para las poblaciones microbianas raras.
El presente estudio utilizó la técnica de genómica unicelular para analizar los metagenomas bacterianos de 10 muestras de koumiss colectadas de Mongolia y Mongolia Interior de China. El trabajo actual ha aplicado tecnologías de vanguardia en la investigación de la diversidad bacteriana en los productos lácteos. Nuestro trabajo ha demostrado la viabilidad de descubrir taxones de baja abundancia mediante la aplicación del enfoque metagenómico de una sola célula. Los resultados alentadores promoverían el desarrollo y la aplicación de enfoques novedosos para abordar problemas en un campo de investigación tradicional.
Materiales y métodos
Preparación de muestras
Se recogieron un total de 10 muestras de koumiss de Mongolia (MG14, MG15, MG16, MG17 y MG18) y Mongolia Interior de China (NM17, NM18, NM19, NM20 y NM21) para el estudio metagenómico. Las muestras se recogieron de forma aséptica y se transportaron en hielo seco.
Se pretrató un mililitro de cada muestra de acuerdo con la metodología descrita en Ward et al. (2013) con algunas modificaciones. Brevemente, las muestras se descongelaron en un baño de hielo durante 3-5 minutos. Después de que las muestras se fundieron, fueron sometidas a centrifugación a baja velocidad para eliminar impurezas y grumos de células eucariotas. Las células procariotas se pelaron de los sueros de leche mediante centrifugación a 13.000 × g durante 15 minutos. Los pellets se resuspendieron en solución salina tamponada con fosfato (PBS) de 2 mL con Tritón X-100 al 1% y se incubaron durante 2 h a 37°C para lisar las células eucariotas restantes. Posteriormente, las bacterias se granularon mediante centrifugación a 13.000 × g durante 15 minutos y los gránulos se volvieron a suspender en 500 µL de PBS. Finalmente, el paso de centrifugación se repitió una vez más para lavar las células bacterianas.
Dilución de gradiente y Amplificación de Desplazamiento múltiple
Para detectar las bacterias poco abundantes, la suspensión bacteriana derivada de cada muestra de koumiss se diluyó en serie para una reacción de amplificación posterior. El número de células en cada muestra se estimó aproximadamente bajo un microscopio (Nikon, Tokio, Japón) utilizando una cámara de conteo de células (Qiujing, Shanghai, China). La etapa de dilución se continuó hasta que el número de células en cada suspensión bacteriana alcanzó aproximadamente 100. La amplificación de desplazamiento múltiple de las células diluidas se realizó utilizando el Kit Monocelular REPLI-g (Qiagen, Germantown, MD, EE. UU.) de acuerdo con las instrucciones del fabricante.
Construcción y Secuenciación de bibliotecas
El ADN amplificado fue esquilado aleatoriamente, y se seleccionaron fragmentos de aproximadamente 500 pb. Después de la construcción de la biblioteca, se utilizó el sistema de PCR en tiempo Real PerkinElmer LabChip® GX Touch y StepOnePlusTM para la inspección de la calidad de la biblioteca. Por último, se secuenciaron lecturas de extremos emparejados de 125 bp en la plataforma Illumina HiSeq 2500 de acuerdo con las instrucciones del fabricante.
Análisis de datos
Comprobación de calidad de secuencia y Filtrado
Las lecturas sin procesar generadas por el secuenciador pueden contener lecturas artificiales de contaminación del adaptador durante la construcción de la biblioteca. Por lo tanto, se realizaron tres pasos para obtener un conjunto de datos de lectura limpia de alta calidad: (1) eliminación de las lecturas causadas por la contaminación del adaptador; (2) eliminación de lecturas con una puntuación media por debajo de una puntuación phred de Q30, que se consideró el punto de corte más bajo para una base de alta calidad; (3) eliminación de lecturas con un exceso significativo de «N» (≥5% de la lectura). El análisis posterior se basó en los datos limpios. Además, se calculó la calidad de la base estadística, basada en el Q30 y el contenido de CG.
Se realizaron alineaciones contra el genoma del huésped para eliminar las secuencias de contaminantes originadas por el huésped. Cualquier lectura originada por el huésped se descartó antes de una comparación adicional con secuencias de referencia del genoma de bacterias (o virus). Para obtener resultados más precisos, en las alineaciones se utilizó el modelo MEM Burrows–Wheeler (BWA) (Versión 0.97 a) (Li y Durbin, 2009).
Asignación taxonómica y Diversidad
El software web Metaphlan se utilizó para la asignación taxonómica a niveles de género y especie (Segata et al., 2012). Para comparar la diversidad de especies dentro de las muestras y entre ellas, analizamos la diversidad alfa y beta por el paquete relacionado con R.
Ensamblaje leído, Predicción de genes y Anotación
Para obtener información más completa, ensamblamos los fragmentos cortados en genoma (contigs). Sin embargo, debido a la presencia de múltiples especies, que es común en muestras metagenómicas, mejoramos el método de ensamblaje del genoma bioinformático que normalmente se usa para el análisis de especies individuales mediante la integración de PICAS (Versión 3.6.2), scripts internos y bases de datos metagenómicas (Zerbino y Birney, 2008; Nurk et al., 2013).
Se utilizó el software MetaGeneMark para la predicción génica de los contig ensamblados (Noguchi et al., 2006). Los genes redundantes se eliminaron mediante CD-HIT con una cobertura de identidad del 90 y el 95% (Li y Godzik, 2006; Fu et al., 2012). Las abundancias relativas de los genes se determinaron alineando lecturas de secuenciación de alta calidad con el catálogo de genes utilizando el mismo procedimiento. El análisis discrepante aguas abajo se basó en la abundancia relativa de genes. La anotación de genes se realizó alineando las secuencias de alta calidad con varias bases de datos públicas (a saber, base de datos no redundante NCBI, NR; Grupos de Grupos Ortólogos de proteínas, COGs; Kyoto Encyclopedia of Genes and Genomes, KEGG) usando BLAST (Altschul et al., 1997). Se realizó una búsqueda de dominios mediante Interproscan (Mulder y Apweiler, 2008).
Números de adhesión de Secuencias de nucleótidos
Los datos de secuencia notificados en este estudio se han depositado en la base de datos SRA (Número de adhesión.SRP083102).
Resultados
Diseño experimental y secuenciación
Para detectar las bacterias poco abundantes en el koumiss, se utilizó la técnica de amplificación unicelular para analizar los metagenomas de las muestras. Se obtuvieron tres suspensiones bacterianas, cada una de aproximadamente 100 células, de una muestra independiente de koumiss por dilución en serie. Se analizaron un total de 30 suspensiones diluidas. A cada muestra diluida se le dio un código de muestra diferente, es decir, el número de identidad de la muestra seguido de 1, 2 o 3, que representan las tres diluciones separadas. Sobre la base de la premisa de que algunas especies raras estarán presentes en una de las diluciones, se llevó a cabo una amplificación de desplazamiento múltiple de las células; y se generaron alrededor de 5 Gb de datos para cada suspensión bacteriana koumiss.
Se generaron un total de 1.040.323.864 lecturas en bruto de las 10 muestras de koumiss (un total de 30 suspensiones bacterianas). El número medio de lecturas por cada suspensión de 100 celdas fue de 34.677.462 (Cuadro suplementario S1). Después del recorte y filtrado de las secuencias no calificadas, se obtuvieron 1.018.381.702 lecturas limpias para todas las muestras (Tabla Suplementaria S1). Los valores del índice de Shannon, el índice de Simpson, el índice Chao1 y el número de especies observadas (Figuras 1-4) mostraron que la mayoría de las muestras de koumiss tenían una alta biodiversidad bacteriana. Las curvas de diversidad de Shannon-Wiener mostraron que la profundidad de secuencia era adecuada para todas las muestras (Figura 1).
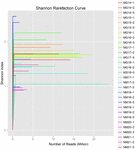
GRÁFICO 1 Curvas de rarefacción de Shannon que estiman la diversidad microbiana de muestras de koumiss.FIGURA
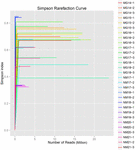
GRÁFICO 2 Curvas de rarefacción de Simpson que estiman la diversidad microbiana de muestras de koumiss.FIGURA
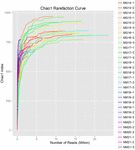
GRÁFICO 3 Curvas de rarefacción de Chao1 que estiman la diversidad microbiana de muestras de koumiss.FIGURA
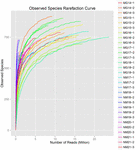
GRÁFICO 4 Índice de rarefacción de especies observadas que estima la diversidad microbiana de muestras de koumiss.
Anotación taxonómica
Las secuencias de alta calidad se asignaron a diferentes niveles taxonómicos para permitir un análisis en profundidad de las comunidades bacterianas de la muestra. Con referencia a algunos estudios publicados sobre la biodiversidad koumiss (Wu et al., 2009; Hao et al., 2010; Sun et al., 2010), clasificamos las bacterias asociadas al koumiss conocidas y no notificadas previamente como taxones comunes y raros, respectivamente.
Las secuencias de alta calidad representaban 13 géneros diferentes (Figura 5). Tres de ellos tenían una abundancia relativa promedio de más del 1%, incluidos Lactobacillus (L.), Lactococcus y Estreptococos. Particularmente, Lactobacillus y Lactococcus fueron los dos géneros más abundantes encontrados en el koumiss. La proporción de lactobacilos en las muestras osciló entre el 52,72 y el 99,96%. Dos miembros de este género, L. helveticus y L. kefiranofaciens, fueron dominados entre la mayoría de las muestras de koumiss (Figura 6). La especie L. buchneri estaba presente principalmente en las muestras de Mongolia, mientras que Lactococcus lactis se detectó en la mayoría de las muestras independientemente de la región de muestreo.
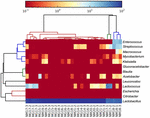
GRÁFICO 5 Mapa de calor que muestra la abundancia relativa de bacterias detectadas en las muestras de koumiss a nivel de género.FIGURA
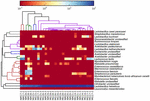
GRÁFICO 6 Mapa de calor que muestra la abundancia relativa de bacterias detectadas en las muestras de koumiss a nivel de especie.
Ensamblaje Metagenómico, Predicción Génica y Anotación Funcional
El ensamblaje de las lecturas resultó en una longitud de ensamblaje de 614,392,623 pb. Los valores de N50 para los conjuntos oscilaron entre 5.596 y 35.200 pa (Tabla suplementaria S2). El número de genes predichos en las muestras de koumiss varió de 10.347 a 34.547, con una longitud promedio de 647,82 a 985,33 pa (Tabla suplementaria S3). Aunque los análisis funcionales empíricos de genes estaban más allá del alcance del estudio actual, anotamos los microbiomas bacterianos koumiss utilizando las bases de datos COG y KEGG, que predijeron la función génica en gran medida basada en la homología de secuencias.
Un total de 545 coincidencias en la salida de anotación mostraron una alta homología a los genes de utilización de lactosa (categoría de dientes de carbohidratos y metabolismo, G), y algunos de ellos se ubicaron dentro del mismo contig (Tabla 1).
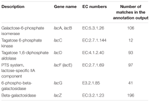
CUADRO 1 Genes relacionados con el metabolismo de la lactosa anotados en el metagenoma bacteriano koumiss.
Algunas otras secuencias podrían codificar genes putativos dentro de la categoría de engranajes de transporte y metabolismo de aminoácidos (E), incluidas secuencias que correspondían a proteinasas que degradan la caseína, el sistema Opp para tomar oligopéptidos de 4-18 residuos y aminopeptidasas (por ejemplo, leucil aminopeptidasa, peptidil-dipeptidasa A, aminopeptidasa N, prolina iminopeptidasa, y endopeptidasa). Aunque algunas secuencias compartieron altas similitudes con las aminotransferasas específicas para arginina, aspartato, metionina e isoleucina, solo se identificó una coincidencia significativa, que correspondía a un supuesto dominio de clase I/clase II originado por S. macedonicus (IPR004839) que contenía aminotransferasas específicas para tirosina y fenilalanina. Finalmente, varias secuencias compartieron una alta homología con las aminoácidos liasas, incluyendo S-ribosilhomocisteína liasa, argininosuccinato liasa, aspartato amoníaco-liasa, cistationina gamma-liasa, histidina amoníaco-liasa y O-acetilhomoserina (tiol)-liasa.
Discusión
El Koumiss es un popular producto lácteo fermentado tradicional en Mongolia y Mongolia Interior de China. Aunque se han realizado varios estudios para investigar la diversidad bacteriana en el koumiss, se ha obtenido poca información sobre la capacidad genética de la microbiota koumiss. Aquí, aplicamos la técnica de amplificación unicelular para analizar los metagenomas bacterianos koumiss, centrándose particularmente en la población bacteriana de baja abundancia.
El enfoque metagenómico típico se ha aplicado previamente para describir la microbiota koumiss. Sin embargo, debido al alto costo de lograr una secuencia profunda, la población de microbiota rara en las muestras a menudo se cubre de manera inadecuada. Por lo tanto, los metagenomas filogénicos y funcionales de las bacterias minoritarias permanecen limitados. Nuestro método de amplificación de una sola célula implicaba la dilución en serie de muestras a suspensiones de 100 células. Debido al bajo número de células presentes en las muestras diluidas de koumiss, sólo se extraería una cantidad limitada de ADN. El paso de amplificación aumentó la cantidad de materiales de ADN a analizar y, por lo tanto, facilitó el análisis de metagenomas de muestras que contenían cantidades de ADN diminutas. Un inconveniente de este método fue la dificultad para garantizar que las secuencias de cada taxón se amplificaran por igual en el proceso; por lo tanto, los resultados obtenidos aquí solo podrían reflejar las abundancias relativas de secuencias, pero no las cantidades absolutas de taxones o genes funcionales identificados. Sin embargo, esto tendría poco efecto en nuestro análisis, ya que el presente estudio difiere de otros trabajos publicados en el enfoque de la población bacteriana rara. Los datos generados por este trabajo proporcionan información complementaria a la población subrepresentada. Creemos que el enfoque actual es adecuado para el análisis futuro de la diversidad de bacterias para diferentes tipos de entornos ecológicos.
La microbiota bacteriana koumiss se compone principalmente de bacterias de ácido láctico (LABORATORIO) y algunas bacterias de ácido acético (Zhang y Zhang, 2011). Como era de esperar, nuestro conjunto de datos contenía principalmente secuencias que representaban el LABORATORIO y las bacterias de ácido acético. Las secuencias correspondientes a Lactobacillus helveticus predominaron en todas las muestras. Además, secuencias que representan a la especie L. kefiranofaciens. L. buchneri. L. kefiranofaciens. Enterococcus (E.) casseliflavus. E. faecalis. E. faecium. Leuconostoc mesenteroides. También se encontraron Lactococcus lactis y Acetobacter pasteurianus. La identificación de secuencias de diferentes taxones puede no ser suficiente para mostrar la viabilidad de las bacterias, sin embargo, refleja la comunidad bacteriana en algún momento del proceso de fermentación. Miyamoto et al. (2010) sugiere que la prevalencia bacteriana en los productos fermentados finales está relacionada con su tolerancia al estrés ácido. En general, los lactobacilos tienen una mayor tolerancia a los ácidos que los lactococos, lo que puede explicar la alta abundancia relativa de secuencias de lactobacilos presentes en nuestro conjunto de datos. Por otro lado, la frecuente aparición de L. helveticus en koumiss coincidió con la observación de la dominancia de las secuencias de L. helveticus.
En nuestro conjunto de datos, algunas secuencias correspondían a L. otakiensis, que es una especie rara que nunca se ha reportado en koumiss u otros nichos lácteos. La especie se describió en primer lugar y se aisló de la solución de decapado no salada utilizada en la producción de sunki, un encurtido japonés tradicional (Watanabe et al., 2009). Fue descubierto por un perfil de polimorfismo de longitud de fragmento amplificado basado en el gen recA. Desde entonces, no se ha informado de que esta especie esté asociada con otros nichos relacionados con los alimentos. Por lo tanto, es probable que esta bacteria pertenezca a la flora autóctona de los encurtidos. Sin embargo, no podemos excluir la posibilidad de que no se haya encontrado simplemente debido a la sensibilidad del método de detección empleado. Lactobacillus otakiensis puede producir aminoácidos de cadena ramificada d, como d-leucina, d-allo-isoleucina y d-valina (Doi et al., 2013). Tiene potencial para ser utilizado en la mejora de las características de producción de ciertos alimentos fermentados(Kato et al., 2011).
Nuestro estudio también identificó secuencias que representan a la especie S. macedonicus, que nunca se ha reportado como una bacteria asociada al koumiss. En cambio, es un cultivo inicial presente en los quesos griegos de oveja y cabra (Georgalaki et al., 2000). Algunos miembros de esta especie son capaces de producir exopolisacáridos, bacteriocinas (Vincent et al., 2001; Anastasiou et al., 2015), y ácido gamma-aminobutírico (Franciosi et al., 2015). A pesar de que esta especie se aísla con frecuencia de alimentos fermentados, el nicho original de S. macedonicus ha sido controvertido (Guarcello et al., 2016). No hasta hace poco, Papadimitriou et al. (2015) identificaron un plásmido adquirido, pSMA198, de Lactococcus lactis. El plásmido probablemente se transfirió a través de un evento de intercambio genético ancestral dentro de un entorno de productos lácteos, dando a entender el origen lácteo de S. macedonicus (Papadimitriou et al., 2015). Similar a S. thermophilus. S. macedonicus está estrechamente relacionado con los comensales y patógenos oportunistas del complejo S. bovis/S. equinus.
La identificación de secuencias que representan los pares de Ruminococcus de la especie fue inesperada, ya que esta bacteria generalmente se encuentra en el ambiente intestinal. Es un microbio intestinal humano normal que puede degradar oligosacáridos de mucina mediante la producción constitutiva de glucosidasas secretoras (Hoskins et al., 1985). Evidencia clínica reciente muestra que la abundancia fecal de esta especie está alterada en niños con trastorno del espectro autista; sin embargo, su papel en el trastorno sigue sin estar claro (Wang et al., 2013).
Sorprendentemente, algunas de las secuencias representaban patógenos potenciales. Por ejemplo, la neumonía por Klebsiella es un patógeno humano oportunista que reside en alrededor del 40% de las tripas humanas y animales. Mycobacterium orygis, anteriormente llamado bacilo oryx, es un miembro del complejo Mycobacterium tuberculosis que puede causar tuberculosis humana (Dawson et al., 2012). Estas dos especies han sido reportadas en leche cruda, pero no en koumiss. Por lo tanto, su presencia podría atribuirse a la contaminación durante la producción convencional de koumiss, especialmente en condiciones de manipulación no aséptica o de baja asepsia.
El metabolismo bacteriano juega un papel importante en la formación de las características y la calidad del koumiss. Se requieren procesos basados en microbios, como la lipólisis y la proteólisis, para sintetizar compuestos aromáticos y aromáticos de koumiss (Gesudu et al., 2016). Consistentemente, el metagenoma bacteriano contenía secuencias que potencialmente codifican la degradación de la lactosa y los sistemas proteolíticos. A diferencia de las vías catabólicas de la lactosa relativamente simples, los sistemas proteolíticos de LABORATORIO se componen de una amplia gama de enzimas (Chen et al., 2014). En comparación con otros LABORATORIOS koumiss detectados, la especie dominante L. helveticus se caracteriza por una alta actividad proteolítica (Zhang et al., 2015). La mayoría de los LABORATORIOS poseen solo una proteinasa de envoltura celular que inicia la hidrólisis de la caseína de la leche, mientras que L. helveticus contiene al menos dos de estas enzimas, a saber, PrtH y PrtH2 (Zhao et al., 2011). Por lo tanto, las altas proporciones de secuencias correspondientes a las especies proteolíticas fuertes de L. helveticus y los genes relacionados con la proteólisis pueden vincularse al contenido relativamente alto de péptidos y aminoácidos libres en el koumiss.
Una dificultad en la producción de koumiss a escala industrial es el control de la percepción del sabor, ya que el koumiss se ha elaborado tradicionalmente mediante fermentación natural. Por lo tanto, ha sido difícil definir el sabor y el perfil de los componentes clave del sabor, particularmente en presencia de contaminantes naturales. La producción de componentes clave de sabor es el resultado de la degradación fermentativa y enzimática de aminoácidos, como los aminoácidos de cadena ramificada, la metionina y los aminoácidos aromáticos (Ardo, 2006). Ejemplos de componentes de sabor incluyen aldehídos, ácidos orgánicos y ésteres, que están formados por la vía de las transaminasas (AT) (Helinck et al., 2004). Esta vía es iniciada por una transaminasa que cataliza la conversión de un aminoácido en su ácido α-ceto correspondiente (Helinck et al., 2004). Algunas secuencias de nuestro conjunto de datos correspondían a aminoácidos de cadena ramificada y aminotransferasas aromáticas. En particular, encontramos un supuesto aminoácido aromático aminotransferasa I en el contig de la especie asociada al koumiss, S. macedonicus. Dado que nuestro trabajo actual solo anotó el microbioma in silico, nuestros datos no son suficientes para mostrar que estas secuencias genéticas identificadas eran realmente funcionales para producir los compuestos de sabor koumiss antes mencionados. La presencia de estos genes, sin embargo, sugiere que son algunos posibles candidatos para tales actividades fermentativas; sin embargo, se requeriría más trabajo experimental para dilucidar sus funciones funcionales exactas.
Además, encontramos secuencias que corresponden a otras vías de conversión de aminoácidos, incluidas las liasas de aminoácidos y la treonina aldolasa. La enzima anterior cataliza la conversión de metionina en metanotiol (Irmler et al., 2008), lo que resulta en la formación de disulfuro de dimetilo y trisulfuro de dimetilo (Fernandez et al., 2000), mientras que este último cataliza la conversión de treonina en glicina y acetaldehído (Ott et al., 2000). De manera similar, la mera localización de estas secuencias dentro del microbioma koumiss no puede servir como evidencia definitiva de sus funciones biológicas reales; se requerirá confirmación experimental futura.
Conclusión
El presente estudio utilizó un método metagenómico modificado para analizar el microbioma bacteriano de muestras de koumiss colectadas de Mongolia y Mongolia Interior de China. Caracterizamos los metagenomas filogénicos y funcionales de las especies raras en el koumiss; y nuestro conjunto de datos refleja rasgos que son de interés y potencial biotecnológico. Nuestro estudio ha demostrado por primera vez la viabilidad de incorporar las técnicas de amplificación unicelular para detectar la microbiota bacteriana koumiss, así como los contaminantes microbianos. Las técnicas desarrolladas en este documento se pueden utilizar en estudios futuros para monitorear los cambios en el microbioma koumiss a lo largo del proceso de fermentación, con enfoque en la población microbiana minoritaria. Además, otras técnicas omicas, como la transcriptómica, la metabolómica, se pueden acoplar al análisis metagenómico actual para confirmar las funciones y la capacidad metabólica del microbioma koumiss.
Técnicamente, el siguiente paso de este trabajo sería optimizar el método actual. Por ejemplo, al aumentar la dilución de la muestra antes de la amplificación del ADN, se puede mejorar la posibilidad de descubrir bacterias raras y novedosas. Por otro lado, se puede emplear una técnica de secuenciación alternativa que puede generar lecturas largas, como la tecnología de secuenciación en tiempo real de una sola molécula de Pacific Biosciences, para mejorar el proceso de ensamblaje del genoma.
Contribuciones de los autores
WZ y HZ diseñaron el estudio. WZ, GY, JY y L-YK escribieron el manuscrito. QH, WH, WL, BM y TS realizaron experimentos. Datos analizados por WZ y QH. Todos los autores revisaron el manuscrito.
Declaración de Conflicto de Intereses
Los autores declaran que la investigación se realizó en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un conflicto de intereses potencial.
Reconocimiento
Esta investigación fue apoyada por la Fundación Nacional de Ciencias Naturales de China (Subvención No. 31571815).
Material suplementario
El Material Suplementario para este artículo se puede encontrar en línea en: https://www.frontiersin.org/article/10.3389/fmicb.2017.00165/full#supplementary-material
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST y PSI-BLAST: una nueva generación de programas de búsqueda de bases de datos de proteínas. Nucleic Acids Res. 25, 3389-3402. doi: 10.1093/nar | 25.17.3389
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Anastasiou, R., Driessche, G. V., Boutou, E., Kazou, M., Alexandraki, V., Vorgias, C. E., et al. (2015). Las cepas de Streptococcus macedonicus dirigidas a un fenotipo osmótico resistente al estrés conservan su capacidad de producir la bacteriocina macedocina en condiciones hiperosmóticas. J. Biotechnol. 212, 125–133. doi: 10.1016 / j. jbiotec.2015.08.018
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Ardo, Y. (2006). Formación de sabor por catabolismo de aminoácidos. Biotechnol. Adv. 24, 238-242. doi: 10.1016 / j.biotechadv.2005.11.005
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
Chen, Y. F., Zhao, W. J., Wu, R. N., Sun, Z. H., Zhang, W. Y., Wang, J.C., et al. (2014). Análisis proteómico de Lactobacillus helveticus H9 durante el crecimiento en leche descremada. J. Dairy Sci. 97, 7413–7425. doi: 10.3168 / jds.2014-8520
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Dawson, K. L., Bell, A., Kawakami, R. P., Coley, K., Yates, G., and Collins, D. M. (2012). Transmission of Mycobacterium orygis (M. tuberculosis complex species) from a tuberculosis patient to a dairy cow in New Zealand. J. Clin. Microbiol. 50, 3136–3138. doi: 10.1128 / JCM.01652-12
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
Doi, K., Mori, K., Mutaguchi, Y., Tashiro, K., Fujino, Y., Ohmori, T., et al. (2013). Proyecto de secuencia del genoma del productor de aminoácidos de cadena ramificada D Lactobacillus otakiensis JCM 15040T, aislado de un pepinillo japonés tradicional. Anuncio del Genoma. 1, e546-e513. doi: 10.1128 / genomeA.00546-13
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
Fernández, M., van Doesburg, W., Rutten, G. A., Marugg, J. D., Alting, A.C., van Kranenburg, R., et al. (2000). Análisis molecular y funcional del gen metC de Lactococcus lactis, codificador de la cistationina beta-liasa. Appl. Environ. Microbiol. 66, 42–48. doi: 10.1128 / AEM.66.1.42-48.2000
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Franciosi, E., Carafa, I., Nardin, T., Schiavon, S., Poznanski, E., Cavazza, A., et al. (2015). Biodiversidad y producción de ácido gamma-aminobutírico por bacterias de ácido láctico aisladas de quesos tradicionales alpinos de leche cruda de vaca. Biomed Res. Int. 2015, 625740. doi: 10.1155/2015/625740
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: acelerado para agrupar los datos de secuenciación de próxima generación. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
PubMed Abstract | CrossRef Full Text | Google Scholar
Georgalaki, M. D., Sarantinopoulos, P., Ferreira, E. S., De Vuyst, L., Kalantzopoulos, G., and Tsakalidou, E. (2000). Biochemical properties of Streptococcus macedonicus strains isolated from Greek Kasseri cheese. J. Appl. Microbiol. 88, 817–825. doi: 10.1046/j.1365-2672.2000.01055.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Gesudu, Q. M., Zheng, Y., Xi, X. X., Hou, Q. C., Xie, H. Y., Huang, W. Q., et al. (2016). Investigating bacterial population structure and dynamics in traditional koumiss from Inner Mongolia using single molecule real-time sequencing. J. Dairy Sci. 99, 7852–7863. doi: 10.3168 / jds.2016-11167
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
Guarcello, R., Carpino, S., Gaglio, R., Pino, A., Rapisarda, T., Caggia, C., et al. (2016). Una aplicación a gran escala de fábrica de bacterias lácticas autóctonas seleccionadas para la producción de queso DOP Pecorino Siciliano. Microbiol de los alimentos. 59, 66–75. doi: 10.1016 / j. fm. 2016.05.011
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Hao, Y., Zhao, L., Zhang, H., Zhai, Z., Huang, Y., Liu, X., et al. (2010). Identificación de la biodiversidad bacteriana en el koumiss mediante electroforesis en gel de gradiente de desnaturalización y reacción en cadena de la polimerasa específica de la especie. J. Dairy Sci. 93, 1926–1933. doi: 10.3168 / jds.2009-2822
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
Helinck, S., Le Bars, D., Moreau, D., and Yvon, M. (2004). Capacidad de las bacterias de ácido láctico termofílico para producir compuestos aromáticos a partir de aminoácidos. Appl. Environ. Microbiol. 70, 3855–3861. doi: 10.1128 / AEM.70.7.3855-3861.2004
Resumen de PubMed | Texto completo de CrossRef / Google Scholar
Hoskins, L. C., Agustines, M., McKee, W. B., Boulding, E. T., Kriaris, M., and Niedermeyer, G. (1985). Degradación de mucina en ecosistemas de colon humano. Aislamiento y propiedades de cepas fecales que degradan antígenos y oligosacáridos del grupo sanguíneo ABH a partir de glicoproteínas de mucina. J. Clin. Invertir. 75, 944–953. doi: 10.1172 | JCI111795
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Irmler, S., Raboud, S., Beisert, B., Rauhut, D., and Berthoud, H. (2008). Clonación y caracterización de dos genes Lactobacillus casei que codifican una cistationina liasa. Appl. Environ. Microbiol. 74, 99–106. doi: 10.1128 / AEM.00745-07
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
Jagielski, V. (1877). El valor del koumiss en el tratamiento de las náuseas, los vómitos y la incapacidad de retener otros alimentos en el estómago. Br. Mediterráneo. J. 2, 919-921. doi: 10.1136 / bmj.2.887.919
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
Kato, S., Ishihara, T., Hemmi, H., Kobayashi, H., and Yoshimura, T. (2011). Alteraciones en las concentraciones de aminoácidos D y en las estructuras microbianas de la comunidad durante la fermentación de vinos tintos y blancos. J. Biosci. Bioeng. 111, 104–108. doi: 10.1016 / j. jbiosc.2010.08.019
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Li, H., y Durbin, R. (2009). Alineación rápida y precisa de lectura corta con la transformación Burrows-Wheeler. Bioinformatics 25, 1754-1760. doi: 10.1093 / bioinformática / btp324
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Li, W., y Godzik, A. (2006). Cd-hit: un programa rápido para agrupar y comparar grandes conjuntos de secuencias de proteínas o nucleótidos. Bioinformática 22, 1658-1659. doi: 10.1093 / bioinformática / btl158
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Miyamoto, M., Seto, T., Nakajima, H., Burenjargal, S., Gombojav, A., Demberei, S., et al. (2010). Análisis de electroforesis en gel de gradiente de desnaturalización de bacterias y levaduras de ácido láctico en leche fermentada tradicional de Mongolia. Ciencia de Alimentos. Technol. Res. 16, 319 a 326. doi: 10.3136 / fstr.16.319
Texto completo de CrossRef / Google Scholar
Mulder, N. J., y Apweiler, R. (2008). La base de datos InterPro y herramientas para el análisis de dominios proteicos. Curr. Protocolo. Bioinformática Capítulo 2, Unidad 2.7. doi: 10.1002 / 0471250953. bi0207s21
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Noguchi, H., Park, J., y Takagi, T. (2006). MetaGene: hallazgo de genes procarióticos de secuencias de escopeta del genoma ambiental. Nucleic Acids Res. 34, 5623-5630. doi: 10.1093/nar / gkl723
Resumen de PubMed / Texto completo de CrossRef | Google Scholar
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A. A., Korobeynikov, A., Lapidus, A., et al. (2013). Ensamblaje de genomas unicelulares y mini-metagenomas a partir de productos quiméricos MDA. J. Comput. Biol. 20, 714–737. doi: 10.1089 / cmb.2013.0084
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
Ott, A., Germond, J. E., and Chaintreau, A. (2000). Origen del acetaldehído durante la fermentación de la leche utilizando precursores marcados con (13) C. J. Agric. Química de Alimentos. 48, 1512–1517. doi: 10.1021 / jf9904867
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Pan, D. D., Zeng, X. Q., and Yan, Y. T. (2011). Caracterización de Lactobacillus fermentum SM-7 aislado de koumiss, una bacteria probiótica potencial con efectos reductores del colesterol. J. Sci. Alimento. Agric. 91, 512–518. doi: 10.1002 / jsfa.4214
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
Papadimitriou, K., Anastasiou, R., Maistrou, E., Plakas, T., Papandreou, N. C., Hamodrakas, S. J., et al. (2015). La adquisición mediante transferencia horizontal de genes del plásmido pSMA198 por Streptococcus macedonicus ACA-DC 198 apunta hacia el origen lácteo de la especie. PLoS ONE 10: e0116337. doi: 10.1371 / journal.ponga.0116337
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
Segata, N., Waldron, L., Ballarini, A., Narasimhan, V., Jousson, O., and Huttenhower, C. (2012). Perfiles de comunidades microbianas metagenómicas utilizando genes marcadores específicos de clados únicos. NAT. Methods 9, 811-814. doi: 10.1038 / nmeth.2066
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Sun, Z., Liu, W., Zhang, J., Yu, J., Zhang, W., Cai, C., et al. (2010). Identification and characterization of the dominant lactobacilli isolated from koumiss in China. J. Gén. Appl. Microbiol. 56, 257–265. doi: 10.2323 / jgam.56.257
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
Thompson, J. (1879). El valor del koumiss en las enfermedades debilitantes. Br. Mediterráneo. J. 1, 270. doi: 10.1136 / bmj.1.1466.270-c
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Vincent, S. J., Faber, E. J., Neeser, J. R., Stingele, F., and Kamerling, J. P. (2001). Estructura y propiedades del exopolisacárido producido por Streptococcus macedonicus Sc136. Glycobiology 11, 131-139. doi: 10.1093/glycob / 11.2.131
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Wang, L., Christophersen, C. T., Sorich, M. J., Gerber, J. P., Angley, M. T., and Conlon, M. A. (2013). Aumento de la abundancia de Sutterella spp. y Ruminococcus pares en las heces de niños con trastorno del espectro autista. Mol. Autismo 4, 42. doi: 10.1186/2040-2392-4-42
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
Ward, T. L., Holid, S., Ioshikhes, I., and Altosaar, I. (2013). Metagenoma de leche humana: un análisis de capacidad funcional. Microbiol de BMC. 13:116. doi: 10.1186/1471-2180-13-116
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
Watanabe, K., Fujimoto, J., Tomii, Y., Sasamoto, M., Makino, H., Kudo, Y., et al. (2009). Lactobacillus kisonensis sp. nov., Lactobacillus otakiensis sp. nov., Lactobacillus rapi sp. nov. y Lactobacillus sunkii sp. nov., heterofermentative species isolated from sunki, a traditional Japanese pickle. Int. J. Syst. Evol. Microbiol. 59 (Pt 4), 754-760. doi: 10.1099 / ijs.0.004689-0
Resumen de PubMed | Texto completo de CrossRef / Google Scholar
Wu, R., Wang, L., Wang, J., Li, H., Menghe, B., Wu, J., et al. (2009). Isolation and preliminary probiotic selection of lactobacilli from koumiss in Inner Mongolia. J. Microbiol Básico. 49, 318–326. doi: 10.1002/empleom.200800047
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
Zerbino, D. R., and Birney, E. (2008). Velvet: algoritmos para ensamblado de lectura corta de novo utilizando gráficos de Bruijn. Genome Res. 18, 821-829. doi: 10.1101 / gr.074492.107
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
Zhang, W., Sun, Z., Sun, T., and Zhang, H. (2010a). Cribado por PCR y análisis de secuencias de clústeres de lio en cepas de Lactobacillus casei aisladas de koumiss. Folia Microbiol. (Praha) 55, 603-606. doi: 10.1007 / s12223-010-0097-3
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
Zhang, W., Yu, D., Sun, Z., Wu, R., Chen, X., Chen, W., et al. (2010b). Secuencia completa del genoma de Lactobacillus casei Zhang, una nueva cepa probiótica aislada de koumiss casero tradicional en Mongolia Interior, China. J. Bacteriol. 192, 5268–5269. doi: 10.1128 / JB.00802-10
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
Zhang, W., y Zhang, H. (2011). «Fermentation and koumiss,» in Handbook of Food and Beverage Fermentation Technology, 2nd Edn, ed. Y. H. Hui (Boca Raton, FL: CRC Press), 165-172.
Google Scholar
Zhang, W. Y., Chen, Y. F., Zhao, W. J., Kwok, L. Y., and Zhang, H. P. (2015). Expresión génica del sistema proteolítico de Lactobacillus helveticus H9 durante la fermentación de la leche. Ana. Microbiol. 65, 1171–1175. doi: 10.3168 / jds.2014-8520
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Zhao, W., Chen, Y., Sun, Z., Wang, J., Zhou, Z., Sun, T., et al. (2011). Secuencia completa del genoma de Lactobacillus helveticus H10. J. Bacteriol. 193, 2666–2667. doi: 10.1128 / JB.00166-11
Resumen de PubMed / Texto completo Cruzado / Google Scholar