- Introduction
- Matériaux et méthodes
- Préparation d’échantillons
- Dilution par gradient et amplification à déplacements multiples
- Construction et séquençage de la bibliothèque
- Analyse des données
- Contrôle et filtrage de la qualité des séquences
- Affectation taxonomique et diversité
- Lire l’Assemblage, la Prédiction des gènes et l’Annotation
- Numéros d’Accession de séquence nucléotidique
- Résultats
- Conception expérimentale et séquençage
- Annotation taxonomique
- Assemblage métagénomique, Prédiction génique et Annotation fonctionnelle
- Discussion
- Conclusion
- Contributions des auteurs
- Déclaration de conflit d’intérêts
- Reconnaissance
- Matériel supplémentaire
Introduction
Le Koumiss, également appelé chige, chigo, arrag ou airag, en langue mongole, est un type de produit laitier fermenté traditionnel. C’est un aliment populaire en Mongolie et en Mongolie intérieure de Chine depuis des siècles (Zhang et Zhang, 2011). Les habitants de ces régions consommaient du koumiss lors des grandes festivités et des offrandes sacrificielles (Zhang et Zhang, 2011). La plus ancienne trace de production de koumiss remonte à la dynastie Han (202 av. J.-C.-202 av.J.-C.). Ce produit avait acquis une grande popularité pendant la dynastie Yuan (AD1271-AD1368) (Zhang et al., 2010b). De nos jours, le koumiss est un aliment courant pour les populations locales de Mongolie et de Mongolie-intérieure, bien que seulement dans quelques-unes de ces régions, il soit fabriqué à l’échelle industrielle. Le koumiss fournit non seulement des nutriments riches, y compris des teneurs élevées en acides aminés essentiels et en vitamines, mais on pense également qu’il soulage un large éventail de conditions médicales et qu’il est bénéfique pour les soins postopératoires (Jagielski, 1877; Thompson, 1879).
Traditionnellement, le koumiss est généralement produit dans des fûts en bois, des récipients en peau d’animal ou des urnes. La fermentation se produit naturellement à température ambiante après l’ajout de lait de jument filtré dans le récipient avec du vieux koumiss, qui sert de culture de départ (Zhang et Zhang, 2011). Le koumiss est une bonne source de nouvelles bactéries à potentiel biotechnologique (Zhang et al., 2010a; Pan et coll., 2011). Par conséquent, il est d’un intérêt intense d’explorer et de préserver autant de bactéries koumiss associées à la fermentation que possible. Au cours des dernières décennies, un certain nombre d’études ont été réalisées pour étudier la communauté bactérienne de koumiss (Wu et al., 2009; Hao et coll., 2010), principalement étudiées par des méthodes basées sur la culture, la biologie moléculaire et le pyroséquençage (Sun et al., 2010). Parmi ces différentes approches, la méthode basée sur le pyroséquençage a fourni le profil microbiote le plus complet du koumiss indépendamment des traits phénotypiques et des problèmes de cultivabilité des microbes individuels. Cependant, le spectre des gènes fonctionnels codés par la bactérie koumiss et leurs capacités fermentatives restent mal caractérisés, en particulier pour les populations microbiennes rares.
La présente étude a utilisé la technique de génomique unicellulaire pour analyser les métagénomes bactériens de 10 échantillons de koumiss collectés en Mongolie et en Mongolie intérieure de Chine. Les travaux actuels ont appliqué des technologies de pointe dans l’étude de la diversité bactérienne dans les produits laitiers. Nos travaux ont démontré la faisabilité de découvrir des taxons peu abondants en appliquant l’approche métagénomique unicellulaire. Les résultats encourageants favoriseraient le développement et l’application de nouvelles approches pour résoudre les problèmes dans un domaine de recherche traditionnel.
Matériaux et méthodes
Préparation d’échantillons
Un total de 10 échantillons de koumiss ont été collectés en Mongolie (MG14, MG15, MG16, MG17 et MG18) et en Mongolie intérieure de Chine (NM17, NM18, NM19, NM20 et NM21) pour l’étude métagénomique. Les échantillons ont été prélevés de manière aseptique et transportés dans de la glace carbonique.
Un millilitre de chaque échantillon a été prétraité selon la méthodologie décrite dans Ward et al. (2013) avec quelques modifications. Brièvement, les échantillons ont été décongelés dans un bain de glace pendant 3 à 5 min. Une fois les échantillons fondus, ils ont été soumis à une centrifugation à basse vitesse pour éliminer les impuretés et les amas de cellules eucaryotes. Les cellules procaryotes ont ensuite été granulées à partir des sérums de lait par centrifugation à 13 000 × g pendant 15 min. Les pastilles ont été remises en suspension dans 2 mL de solution saline tamponnée au phosphate (PBS) avec du Triton X-100 à 1% et incubées pendant 2 h à 37°C pour lyser les cellules eucaryotes restantes. Par la suite, les bactéries ont été granulées par centrifugation à 13 000 × g pendant 15 min et les pastilles ont été remises en suspension dans 500 µL de PBS. Enfin, l’étape de centrifugation a été répétée une fois de plus pour laver les cellules bactériennes.
Dilution par gradient et amplification à déplacements multiples
Pour détecter les bactéries peu abondantes, la suspension bactérienne dérivée de chaque échantillon de koumiss a été diluée en série pour une réaction d’amplification ultérieure. Le nombre de cellules de chaque échantillon a été estimé grossièrement au microscope (Nikon, Tokyo, Japon) à l’aide d’une chambre de comptage de cellules (Qiujing, Shanghai, Chine). L’étape de dilution a été poursuivie jusqu’à ce que le nombre de cellules dans chaque suspension bactérienne atteigne environ 100. L’amplification à déplacement multiple des cellules diluées a été réalisée à l’aide du kit Cellule unique REPLI-g (Qiagen, Germantown, MD, USA) selon les instructions du fabricant.
Construction et séquençage de la bibliothèque
L’ADN amplifié a été cisaillé au hasard et les fragments d’environ 500 pb ont été sélectionnés. Après la construction de la bibliothèque, le système de PCR en temps réel LabChip® GX Touch et StepOnePlusTM de PerkinElmer ont été utilisés pour l’inspection de la qualité de la bibliothèque. Enfin, des lectures d’extrémité appariées de 125 pb ont été séquencées sur la plate-forme Illumina HiSeq 2500 selon les instructions du fabricant.
Analyse des données
Contrôle et filtrage de la qualité des séquences
Les lectures brutes générées par le séquenceur peuvent contenir des lectures artificielles de contamination de l’adaptateur pendant la construction de la bibliothèque. Par conséquent, trois étapes ont été effectuées pour obtenir un ensemble de données de lecture propre de haute qualité: (1) élimination des lectures causées par la contamination de l’adaptateur; (2) suppression des lectures avec un score moyen inférieur à un score phred de Q30, qui était considéré comme le seuil le plus bas pour une base de haute qualité; (3) suppression des lectures avec un excès significatif de « N » (≥5% de la lecture). L’analyse en aval était basée sur les données propres. De plus, la qualité de base statistique, basée sur Q30 et la teneur en GC, a été calculée.
Des alignements par rapport au génome de l’hôte ont été effectués pour éliminer les séquences de contaminants provenant de l’hôte. Toutes les lectures provenant de l’hôte ont été éliminées avant une comparaison plus poussée avec les séquences de référence du génome des bactéries (ou des virus). Pour obtenir des résultats plus précis, le modèle MEM de l’aligneur Burrows–Wheeler (BWA) (version 0.97a) a été utilisé dans les alignements (Li et Durbin, 2009).
Affectation taxonomique et diversité
Le logiciel web Metaphlan a été utilisé pour l’affectation taxonomique aux niveaux des genres et des espèces (Segata et al., 2012). Pour comparer la diversité des espèces au sein et entre les échantillons, nous avons analysé la diversité alpha et bêta par le paquet lié à R.
Lire l’Assemblage, la Prédiction des gènes et l’Annotation
Pour obtenir des informations plus complètes, nous avons assemblé les fragments cisaillés en génome (contigs). Cependant, en raison de la présence de plusieurs espèces, ce qui est courant dans les échantillons métagénomiques, nous avons amélioré la méthode d’assemblage du génome bioinformatique normalement utilisée pour l’analyse d’une seule espèce en intégrant des PIQues (version 3.6.2), des scripts internes et des bases de données métagénomiques (Zerbino et Birney, 2008; Nurk et al., 2013).
Le logiciel MetaGeneMark a été utilisé pour la prédiction génétique des contigs assemblés (Noguchi et al., 2006). Les gènes redondants ont été éliminés à l’aide de CD-HIT avec une couverture d’identité de 90 et 95% (Li et Godzik, 2006; Fu et al., 2012). Les abondances relatives des gènes ont été déterminées en alignant des lectures de séquençage de haute qualité au catalogue de gènes en utilisant la même procédure. L’analyse des écarts en aval était basée sur les abondances relatives des gènes. L’annotation des gènes a été réalisée en alignant les séquences de haute qualité sur plusieurs bases de données publiques (à savoir la base de données non redondante NCBI, NR; Grappes de groupes orthologues de protéines, COGs; Encyclopédie des Gènes et des génomes de Kyoto, KEGG) utilisant BLAST (Altschul et al., 1997). Une recherche de domaine a été effectuée à l’aide d’Interproscan (Mulder et Apweiler, 2008).
Numéros d’Accession de séquence nucléotidique
Les données de séquence rapportées dans cette étude ont été déposées dans la base de données SRA (Numéro d’accession.SRP083102).
Résultats
Conception expérimentale et séquençage
Pour détecter les bactéries peu abondantes dans le koumiss, la technique d’amplification unicellulaire a été utilisée pour analyser les métagénomes des échantillons. Trois suspensions bactériennes, chacune d’environ 100 cellules, ont été dérivées d’un échantillon de koumiss indépendant par dilution en série. Au total, 30 suspensions diluées ont été analysées. Chaque échantillon dilué a reçu un code d’échantillon différent, c’est-à-dire le numéro d’identité de l’échantillon suivi de 1, 2 ou 3, représentant les trois dilutions distinctes. En partant du principe que certaines espèces rares seront présentes dans l’une des dilutions, une amplification à déplacement multiple des cellules a été réalisée; et environ 5 Go de données ont été générés pour chaque suspension bactérienne de koumiss.
Un total de 1 040 323 864 lectures brutes ont été générées à partir des 10 échantillons de koumiss (un total de 30 suspensions bactériennes). Le nombre moyen de lectures pour chaque suspension de 100 cellules était de 34 677 462 (Tableau supplémentaire S1). Après rognage et filtrage des séquences non qualifiées, nous avons obtenu 1 018 381 702 lectures propres pour tous les échantillons (Tableau supplémentaire S1). Les valeurs de l’indice de Shannon, de l’indice de Simpson, de l’indice Chao1 et du nombre d’espèces observées (Figures 1 à 4) ont montré que la plupart des échantillons de koumiss présentaient une biodiversité bactérienne élevée. Les courbes de diversité de Shannon-Wiener ont montré que la profondeur de séquence était adéquate pour tous les échantillons (figure 1).
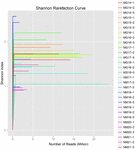
FIGURE 1. Courbes de raréfaction de Shannon qui estiment la diversité microbienne des échantillons de koumiss.
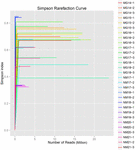
FIGURE 2. Courbes de raréfaction de Simpson qui estiment la diversité microbienne des échantillons de koumiss.
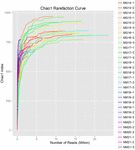
FIGURE 3. Courbes de raréfaction Chao1 qui estiment la diversité microbienne des échantillons de koumiss.
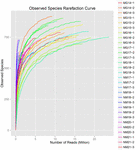
FIGURE 4. Indice de raréfaction des espèces observées qui estime la diversité microbienne des échantillons de koumiss.
Annotation taxonomique
Les séquences de haute qualité ont été affectées à différents niveaux taxonomiques pour permettre une analyse approfondie des communautés bactériennes de l’échantillon. En référence à certaines études publiées sur la biodiversité de koumiss (Wu et al., 2009; Hao et coll., 2010; Sun et coll., 2010), nous avons classé les bactéries associées à koumiss connues et non rapportées précédemment comme taxons communs et rares, respectivement.
Les séquences de haute qualité représentaient 13 genres différents (Figure 5). Trois d’entre eux avaient une abondance relative moyenne de plus de 1%, y compris Lactobacillus (L.), Lactococcus et Streptococcus. En particulier, Lactobacillus et Lactococcus étaient les deux genres les plus abondants trouvés dans le koumiss. La proportion de lactobacilles dans les échantillons variait de 52,72 à 99,96%. Deux membres de ce genre, L. helveticus et L. kefiranofaciens, ont été dominés parmi la plupart des échantillons de koumiss (Figure 6). L’espèce L. buchneri était présent principalement dans les échantillons mongols, tandis que Lactococcus lactis a été détecté dans la plupart des échantillons quelle que soit la région d’échantillonnage.
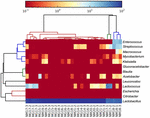
FIGURE 5. Carte thermique montrant l’abondance relative des bactéries détectées dans les échantillons de koumiss au niveau du genre.
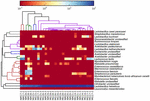
FIGURE 6. Carte thermique montrant l’abondance relative des bactéries détectées dans les échantillons de koumiss au niveau des espèces.
Assemblage métagénomique, Prédiction génique et Annotation fonctionnelle
L’assemblage des lectures a abouti à une longueur d’assemblage de 614 392 623 pb. Les valeurs de N50 pour les assemblages variaient de 5 596 à 35 200 pb (Tableau supplémentaire S2). Le nombre de gènes prédits dans les échantillons de koumiss variait de 10 347 à 34 547, avec une longueur moyenne allant de 647,82 à 985,33 pb (Tableau supplémentaire S3). Bien que les analyses fonctionnelles empiriques des gènes dépassent le cadre de la présente étude, nous avons annoté les microbiomes bactériens de koumiss à l’aide des bases de données COG et KEGG, qui prédisaient la fonction génique en grande partie basée sur l’homologie des séquences.
Un total de 545 correspondances dans la sortie d’annotation ont montré une homologie élevée avec les gènes d’utilisation du lactose (catégorie COGs des glucides et du métabolisme, G), et certains d’entre eux étaient situés dans le même contig (tableau 1).
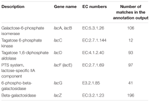
TABLEAU 1. Gènes liés au métabolisme du lactose annotés dans le métagénome bactérien de koumiss.
D’autres séquences pourraient coder des gènes putatifs dans la catégorie COGs du transport et du métabolisme des acides aminés (E), y compris des séquences correspondant à des protéinases dégradant la caséine, le système Opp pour absorber des oligopeptides de 4 à 18 résidus et des aminopeptidases (par exemple, leucyl aminopeptidase, peptidyl-dipeptidase A, aminopeptidase N, proline iminopeptidase , et endopeptidase). Bien que certaines séquences partagent des similitudes élevées avec les aminotransférases spécifiques à l’arginine, à l’aspartate, à la méthionine et à l’isoleucine, une seule correspondance significative a été identifiée, qui correspond à une aminotransférase spécifique à la tyrosine et à la phénylalanine provenant du domaine de classe I/classe II (IPR004839). Enfin, un certain nombre de séquences partageaient une homologie élevée avec les lyases d’acides aminés, notamment la S-ribosylhomocystéine lyase, l’argininosuccinate lyase, l’aspartate ammoniac-lyase, la cystathionine gamma-lyase, l’histidine ammoniac-lyase et l’O-acétylhomosérine (thiol)-lyase.
Discussion
Le koumiss est un produit laitier fermenté traditionnel populaire en Mongolie et en Mongolie intérieure en Chine. Bien qu’un certain nombre d’études aient été réalisées pour étudier la diversité bactérienne dans le koumiss, peu d’informations ont été obtenues concernant la capacité génétique du microbiote du koumiss. Ici, nous avons appliqué la technique d’amplification unicellulaire pour analyser les métagénomes bactériens de koumiss, en nous concentrant particulièrement sur la population bactérienne peu abondante.
L’approche métagénomique typique a déjà été appliquée pour décrire le microbiote de koumiss. Cependant, en raison du coût élevé de la réalisation d’une séquence profonde, la population de microbiote rare dans les échantillons est souvent couverte de manière inadéquate. Ainsi, les métagénomes phylogéniques et fonctionnels des bactéries minoritaires restent limités. Notre méthode d’amplification monocellulaire impliquait la dilution en série d’échantillons en suspensions à 100 cellules. En raison du faible nombre de cellules présentes dans les échantillons de koumiss dilués, seule une quantité limitée d’ADN serait extraite. L’étape d’amplification a augmenté la quantité de matériaux d’ADN à analyser et a donc facilité l’analyse du métagénome d’échantillons contenant des quantités infimes d’ADN. Un inconvénient de cette méthode était la difficulté à s’assurer que les séquences de chaque taxon seraient également amplifiées dans le processus; par conséquent, les résultats obtenus ici ne pouvaient refléter que les abondances relatives de séquences mais pas les quantités absolues de taxons ou de gènes fonctionnels identifiés. Pourtant, cela aurait peu d’effet sur notre analyse car la présente étude diffère des autres travaux publiés en concentrant la population bactérienne rare. Les données générées par ces travaux fournissent des informations complémentaires à la population sous-représentée. Nous pensons que la présente approche convient à l’analyse future de la diversité des bactéries pour différents types d’environnements écologiques.
Le microbiote bactérien de koumiss est principalement composé de bactéries lactiques (LAB) et de certaines bactéries acétiques (Zhang et Zhang, 2011). Comme prévu, notre jeu de données contenait principalement des séquences représentant les bactéries de LABORATOIRE et d’acide acétique. Les séquences correspondant à Lactobacillus helveticus dominaient dans tous les échantillons. Par ailleurs, des séquences représentant l’espèce, L. kefiranofaciens. L. buchneri. L. kefiranofaciens. Enterococcus (E.) casseliflavus. E. faecalis. E. faecium. Leuconostoc mesenteroides. Lactococcus lactis et Acetobacter pasteurianus ont également été trouvés. L’identification de séquences de différents taxons peut ne pas être suffisante pour montrer la viabilité des bactéries, elle reflète néanmoins la communauté bactérienne à un moment donné du processus de fermentation. Miyamoto et coll. (2010) suggèrent que la prévalence bactérienne dans les produits fermentés finaux est liée à leur tolérance au stress acide. Généralement, les lactobacilles ont une tolérance à l’acide plus élevée que les lactocoques, ce qui peut expliquer la forte abondance relative des séquences de lactobacilles présentes dans notre ensemble de données. D’autre part, l’apparition fréquente de L. helveticus à koumiss a coïncidé avec l’observation de la dominance des séquences de L. helveticus.
Dans notre jeu de données, certaines séquences correspondaient à L. otakiensis, qui est une espèce rare qui n’a jamais été signalée dans les koumiss ou d’autres niches laitières. L’espèce a d’abord été décrite et isolée de la solution de décapage non salée utilisée dans la production de sunki, un cornichon japonais traditionnel (Watanabe et al., 2009). Il a été découvert par profilage de polymorphisme de longueur de fragment amplifié basé sur le gène recA. Depuis lors, il n’a pas été signalé que cette espèce soit associée à d’autres niches liées à l’alimentation. Ainsi, il est probable que cette bactérie appartienne à la flore autochtone des cornichons. Cependant, nous ne pouvons exclure la possibilité qu’il n’ait pas été trouvé simplement en raison de la sensibilité de la méthode de détection utilisée. Lactobacillus otakiensis peut produire des acides aminés à chaîne ramifiée en d, tels que la d-leucine, la d-allo-isoleucine et la d-valine (Doi et al., 2013). Il peut être utilisé pour améliorer les caractéristiques de production de certains aliments fermentés (Kato et al., 2011).
Notre étude a également identifié des séquences représentant l’espèce S. macedonicus, qui n’a jamais été signalée comme une bactérie associée à koumiss. Au lieu de cela, il s’agit d’une culture de départ présente dans les fromages grecs de brebis et de chèvre (Georgalaki et al., 2000). Certains membres de cette espèce sont capables de produire des exopolysaccharides, des bactériocines (Vincent et al., 2001; Anastasiou et coll., 2015), et l’acide gamma-aminobutyrique (Franciosi et al., 2015). Même si cette espèce est fréquemment isolée des aliments fermentés, la niche originale de S. macedonicus a été controversée (Guarcello et al., 2016). Pas jusqu’à récemment, Papadimitriou et al. (2015) ont identifié un plasmide acquis, pSMA198, de Lactococcus lactis. Le plasmide a probablement été transféré via un événement d’échange génétique ancestral dans un environnement de produit laitier, ce qui fait allusion à l’origine laitière de S. macedonicus (Papadimitriou et al., 2015). Semblable à S. thermophilus. S. macedonicus est étroitement lié aux commensaux et aux pathogènes opportunistes du complexe S. bovis/S. equinus.
L’identification des séquences représentant les couples de l’espèce Ruminococcus était inattendue, car cette bactérie se trouve généralement dans l’environnement intestinal. C’est un microbe intestinal humain normal qui peut dégrader les oligosaccharides de mucines par la production constitutive de glycosidases sécrétoires (Hoskins et al., 1985). Des preuves cliniques récentes montrent que l’abondance fécale de cette espèce est altérée chez les enfants atteints d’un trouble du spectre autistique; cependant, son rôle dans le trouble reste incertain (Wang et al., 2013).
Étonnamment, certaines des séquences représentaient des agents pathogènes potentiels. Par exemple, la pneumonie à Klebsiella est un agent pathogène humain opportuniste qui réside dans environ 40% des intestins humains et animaux. Mycobacterium orygis, précédemment appelé bacille de l’oryx, est un membre du complexe Mycobacterium tuberculosis qui peut causer la tuberculose humaine (Dawson et al., 2012). Ces deux espèces ont été signalées dans du lait cru mais pas dans du koumiss. Par conséquent, leur présence pourrait être attribuée à la contamination lors de la production conventionnelle de koumiss, en particulier dans des conditions de manipulation non ou peu aseptiques.
Le métabolisme bactérien joue un rôle important dans la formation des caractéristiques et de la qualité du koumiss. Des processus microbiens tels que la lipolyse et la protéolyse sont nécessaires pour synthétiser les composés aromatiques et aromatiques du koumiss (Gesudu et al., 2016). De manière constante, le métagénome bactérien contenait des séquences qui codaient potentiellement pour la dégradation du lactose et les systèmes protéolytiques. Contrairement aux voies cataboliques du lactose relativement simples, les systèmes protéolytiques de LABORATOIRE sont constitués d’une gamme diversifiée d’enzymes (Chen et al., 2014). Par rapport à d’autres laboratoires de koumiss détectés, l’espèce dominante L. helveticus est caractérisée par une activité protéolytique élevée (Zhang et al., 2015). La plupart des LABORATOIRES ne possèdent qu’une seule protéinase de l’enveloppe cellulaire qui déclenche l’hydrolyse de la caséine du lait, alors que L. helveticus contient au moins deux de ces enzymes, à savoir PrtH et PrtH2 (Zhao et al., 2011). Ainsi, les proportions élevées de séquences correspondant aux espèces protéolytiques fortes de L. helveticus et aux gènes liés à la protéolyse peuvent être liées aux teneurs relativement élevées en peptides et en acides aminés libres dans le koumiss.
Une difficulté dans la production industrielle de koumiss est le contrôle de la perception des saveurs, car le koumiss est traditionnellement fabriqué par fermentation naturelle. Ainsi, il a été difficile de définir la saveur et le profil des composants aromatiques clés, en particulier en présence des contaminants naturels. La production de composants aromatiques clés résulte de la dégradation fermentative et enzymatique d’acides aminés, tels que les acides aminés à chaîne ramifiée, la méthionine et les acides aminés aromatiques (Ardo, 2006). Des exemples de composants aromatiques comprennent les aldéhydes, les acides organiques et les esters, qui sont formés par la voie de la transaminase (AT) (Helinck et al., 2004). Cette voie est initiée par une transaminase qui catalyse la conversion d’un acide aminé en son acide α-céto correspondant (Helinck et al., 2004). Certaines séquences de notre jeu de données correspondaient à des aminotransférases à chaîne ramifiée et à acides aminés aromatiques. En particulier, nous avons trouvé un acide aminé aromatique présumé aminotransférase I dans le contig de l’espèce associée à koumiss, S. macedonicus. Comme nos travaux actuels n’ont annoté que le microbiome in silico, nos données ne sont pas suffisantes pour montrer que ces séquences génétiques identifiées étaient effectivement fonctionnelles pour produire les composés aromatiques de koumiss susmentionnés. La présence de ces gènes suggère néanmoins qu’ils sont des candidats possibles pour de telles activités fermentatives; pourtant, d’autres travaux expérimentaux seraient nécessaires pour élucider leurs rôles fonctionnels exacts.
De plus, nous avons trouvé des séquences correspondant à d’autres voies de conversion d’acides aminés, y compris les lyases d’acides aminés et la thréonine aldolase. La première enzyme catalyse la conversion de la méthionine en méthanethiol (Irmler et al., 2008), entraînant ainsi la formation de disulfure de diméthyle et de trisulfure de diméthyle (Fernandez et al., 2000), tandis que cette dernière catalyse la conversion de la thréonine en glycine et en acétaldéhyde (Ott et al., 2000). De même, la simple localisation de ces séquences dans le microbiome de koumiss ne peut pas servir de preuve définitive dans leurs rôles biologiques réels; une confirmation expérimentale future sera nécessaire.
Conclusion
La présente étude a utilisé une méthode métagénomique modifiée pour analyser le microbiome bactérien d’échantillons de koumiss collectés en Mongolie et en Mongolie intérieure en Chine. Nous avons caractérisé à la fois les métagénomes phylogéniques et fonctionnels des espèces rares de koumiss; et notre ensemble de données reflète des traits qui présentent un intérêt et un potentiel biotechnologiques. Notre étude a démontré pour la première fois la faisabilité d’intégrer les techniques d’amplification unicellulaire dans la détection du microbiote bactérien koumiss, ainsi que des contaminants microbiens. Les techniques développées ici peuvent être utilisées dans de futures études pour surveiller les changements dans le microbiome de koumiss tout au long du processus de fermentation, en mettant l’accent sur la population microbienne minoritaire. De plus, d’autres techniques omiques, telles que la transcriptomique, la métabolomique, peuvent être couplées à l’analyse métagénomique actuelle afin de confirmer les fonctions et la capacité métabolique du microbiome de koumiss.
Techniquement, la prochaine étape de ce travail serait d’optimiser la méthode actuelle. Par exemple, en augmentant la dilution de l’échantillon avant l’amplification de l’ADN, les chances de découvrir des bactéries rares et nouvelles peuvent être améliorées. D’autre part, une technique de séquençage alternative pouvant générer de longues lectures, telle que la technologie de séquençage en temps réel à molécule unique de Pacific Biosciences, peut être utilisée pour améliorer le processus d’assemblage du génome.
Contributions des auteurs
WZ et HZ ont conçu l’étude. WZ, GY, JY et L-YK ont écrit le manuscrit. QH, WH, WL, BM et TS ont effectué des expériences. WZ et QH ont analysé les données. Tous les auteurs ont examiné le manuscrit.
Déclaration de conflit d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de relations commerciales ou financières pouvant être interprétées comme un conflit d’intérêts potentiel.
Reconnaissance
Cette recherche a été soutenue par la Fondation Nationale des Sciences naturelles de Chine (Subvention n ° 31571815).
Matériel supplémentaire
Le matériel supplémentaire pour cet article peut être trouvé en ligne à l’adresse suivante:: https://www.frontiersin.org/article/10.3389/fmicb.2017.00165/full#supplementary-material
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et coll. (1997). Gapped BLAST et PSI-BLAST: une nouvelle génération de programmes de recherche de bases de données de protéines. Acides nucléiques Res. 25, 3389-3402. doi: 10.1093/nar/25.17.3389
Résumé PubMed / CrossRef Texte intégral / Google Scholar
Anastasiou, R., Driessche, G. V., Boutou, E., Kazou, M., Alexandraki, V., Vorgias, C. E., et al. (2015). Les souches modifiées de Streptococcus macedonicus vers un phénotype résistant au stress osmotique conservent leur capacité à produire la bactériocine macedocine dans des conditions hyperosmotiques. J. Biotechnol. 212, 125–133. doi: 10.1016 / j.jbiotec.2015.08.018
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Ardo, Y. (2006). Formation de saveur par catabolisme des acides aminés. Biotechnol. Adv. 24, 238-242. doi: 10.1016 / j. biotechadv.2005.11.005
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Chen, Y. F., Zhao, W. J., Wu, R. N., Sun, Z. H., Zhang, W. Y., Wang, J. C., et al. (2014). Analyse du protéome de Lactobacillus helveticus H9 pendant la croissance dans le lait écrémé. J. Dairy Sci. 97, 7413–7425. doi: 10.3168/jds.2014-8520
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Il s’agit de l’un des plus importants ouvrages de l’histoire de l’art et de l’histoire de l’Art. Transmission de Mycobacterium orygis (espèce complexe de M. tuberculosis) d’un patient tuberculeux à une vache laitière en Nouvelle-Zélande. J. Clin. Microbiol. 50, 3136–3138. doi: 10.1128/ JCM.01652-12
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Doi, K., Mori, K., Mutaguchi, Y., Tashiro, K., Fujino, Y., Ohmori, T., et al. (2013). Séquence génomique préliminaire du producteur d’acides aminés à chaîne ramifiée en D Lactobacillus otakiensis JCM 15040T, isolé d’un cornichon japonais traditionnel. Annonce du génome. 1, e546-e513. doi: 10.1128 / genomeA.00546-13
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Fernandez, M., van Doesburg, W., Rutten, G. A., Marugg, J. D., Alting, A. C., van Kranenburg, R., et al. (2000). Analyses moléculaires et fonctionnelles du gène metC de Lactococcus lactis, codant pour la cystathionine bêta-lyase. Appl. Environ. Microbiol. 66, 42–48. doi: 10.1128/AEM.66.1.42-48.2000
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Franciosi, E., Carafa, I., Nardin, T., Schiavon, S., Poznanski, E., Cavazza, A., et al. (2015). Biodiversité et production d’acide gamma-aminobutyrique par des bactéries lactiques isolées des fromages au lait cru de vache alpins traditionnels. Biomed Res. Int. 2015, 625740. doi: 10.1155/2015/625740
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Il y a une grande variété de plantes, de plantes, de plantes, de plantes, de plantes, de plantes, de plantes, de plantes, de plantes, de plantes, de plantes, de plantes, de plantes, de plantes, de plantes, de plantes, de plantes, de plantes, de plantes, etc. CD-HIT : accéléré pour regrouper les données de séquençage de nouvelle génération. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
PubMed Abstract | CrossRef Full Text | Google Scholar
Georgalaki, M. D., Sarantinopoulos, P., Ferreira, E. S., De Vuyst, L., Kalantzopoulos, G., and Tsakalidou, E. (2000). Biochemical properties of Streptococcus macedonicus strains isolated from Greek Kasseri cheese. J. Appl. Microbiol. 88, 817–825. doi: 10.1046/j.1365-2672.2000.01055.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Gesudu, Q. M., Zheng, Y., Xi, X. X., Hou, Q. C., Xie, H. Y., Huang, W. Q., et coll. (2016). Étude de la structure et de la dynamique des populations bactériennes dans le koumiss traditionnel de Mongolie intérieure à l’aide d’un séquençage en temps réel à une seule molécule. J. Dairy Sci. 99, 7852–7863. doi: 10.3168/jds.2016-11167
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Guarcello, R., Carpino, S., Gaglio, R., Pino, A., Rapisarda, T., Caggia, C., et al. (2016). Une application à grande échelle en usine de bactéries lactiques autochtones sélectionnées pour la production de fromage AOP Pecorino Siciliano. Microbiol alimentaire. 59, 66–75. doi: 10.1016 / j.fm.2016.05.011
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Hao, Y., Zhao, L., Zhang, H., Zhai, Z., Huang, Y., Liu, X., et al. (2010). Identification de la biodiversité bactérienne dans le koumiss par électrophorèse sur gel à gradient dénaturant et réaction en chaîne par polymérase spécifique à l’espèce. J. Dairy Sci. 93, 1926–1933. doi: 10.3168/jds.2009-2822
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Le Bar, D., Moreau, D., et Yvon, M. (2004). Capacité des bactéries lactiques thermophiles à produire des composés aromatiques à partir d’acides aminés. Appl. Environ. Microbiol. 70, 3855–3861. doi: 10.1128/AEM.70.7.3855 – 3861.2004
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Il s’agit de l’un des principaux facteurs de croissance de la population. Dégradation de la mucine dans les écosystèmes du côlon humain. Isolement et propriétés des souches fécales qui dégradent les antigènes du groupe sanguin ABH et les oligosaccharides des glycoprotéines de mucine. J. Clin. Investir. 75, 944–953. doi: 10.1172 / JCI111795
Résumé PubMed / Texte intégral Croisé / Google Scholar
Irmler, S., Raboud, S., Beisert, B., Rauhut, D., et Berthoud, H. (2008). Clonage et caractérisation de deux gènes de Lactobacillus casei codant pour une cystathionine lyase. Appl. Environ. Microbiol. 74, 99–106. doi: 10.1128/AEM.00745-07
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Jagielski, V. (1877). La valeur du koumiss dans le traitement des nausées, des vomissements et de l’incapacité de conserver d’autres aliments sur l’estomac. Fr. Med. J. 2, 919-921. doi: 10.1136 / bmj.2.887.919
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Il s’agit d’une espèce de plantes de la famille des » Poaceae « , sous-famille des » pooideae « , sous-famille des » Pooideae « , originaire d’Asie du Sud-Est. Altérations des concentrations d’acides aminés D et des structures de la communauté microbienne lors de la fermentation des vins rouges et blancs. J. Biosci. Bioeng. 111, 104–108. doi: 10.1016/ j.jbiosc.2010.08.019
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Li, H. et Durbin, R. (2009). Alignement rapide et précis en lecture courte avec la transformation Burrows-Wheeler. Bioinformatique 25, 1754-1760. doi: 10.1093/ bioinformatique / btp324
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Li, W. et Godzik, A. (2006). Cd-hit: un programme rapide pour regrouper et comparer de grands ensembles de séquences de protéines ou de nucléotides. Bioinformatique 22, 1658-1659. doi: 10.1093/ bioinformatique / btl158
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Miyamoto, M., Seto, T., Nakajima, H., Burenjargal, S., Gombojav, A., Demberei, S., et al. (2010). Analyse par électrophorèse sur gel à gradient dénaturant de bactéries lactiques et de levures dans le lait fermenté mongol traditionnel. Sci alimentaire. Technol. Rés. 16, 319 à 326. doi: 10.3136/fstr.16.319
CrossRef Texte intégral / Google Scholar
Mulder, N. J., et Apweiler, R. (2008). La base de données InterPro et les outils d’analyse des domaines protéiques. Curr. Protoc. Bioinformatique Chapitre 2, Unit2.7. doi: 10.1002/0471250953.bi0207s21
Résumé PubMed / Texte intégral Croisé / Google Scholar
Noguchi, H., Park, J., et Takagi, T. (2006). MétaGène: procaryotic gene finding from environmental genome shotgun sequences. Acides nucléiques Res. 34, 5623-5630. doi: 10.1093/nar/gkl723
Résumé PubMed / Texte intégral Croisé / Google Scholar
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A. A., Korobeynikov, A., Lapidus, A., et al. (2013). Assemblage de génomes unicellulaires et de mini-métagénomes à partir de produits MDA chimériques. J. Comput. Biol. 20, 714–737. doi: 10.1089/cmb.2013.0084
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Ott, A., Germond, J. E. et Chaintreau, A. (2000). Origine de l’acétaldéhyde lors de la fermentation du lait à l’aide de précurseurs marqués au C (13). J. Agric. Chem alimentaire. 48, 1512–1517. doi: 10.1021 /jf9904867
Résumé PubMed / Texte intégral Croisé / Google Scholar
Il s’agit de la première édition de la série. Caractérisation de Lactobacillus fermentum SM-7 isolé de koumiss, une bactérie probiotique potentielle ayant des effets hypocholestérolémiants. J. Sci. Aliment. Agric. 91, 512–518. doi: 10.1002/jsfa.4214
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Papadimitriou, K., Anastasiou, R., Maistrou, E., Plakas, T., Papandréou, N. C., Hamodrakas, S. J., et al. (2015). L’acquisition par transfert horizontal de gènes du plasmide pSMA198 par Streptococcus macedonicus ACA-DC 198 pointe vers l’origine laitière de l’espèce. PLoS ONE 10: e0116337. doi: 10.1371 / journal.pone.0116337
Résumé PubMed / CrossRef Texte intégral / Google Scholar
Il s’agit d’une espèce de plantes de la famille des » Poaceae « , sous-famille des » pooideae « , sous-famille des » Pooideae « , originaire d’Asie du Sud-Est. Profilage de la communauté microbienne métagénomique à l’aide de gènes marqueurs uniques spécifiques à un clade. NAT. Méthodes 9, 811-814. doi: 10.1038/nmeth.2066
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Sun, Z., Liu, W., Zhang, J., Yu, J., Zhang, W., Cai, C., et al. (2010). Identification et caractérisation des lactobacilles dominants isolés de koumiss en Chine. J. Gen. Appl. Microbiol. 56, 257–265. doi: 10.2323/jgam.56.257
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Thompson, J. (1879). La valeur du koumiss dans le gaspillage des maladies. Fr. Med. J. 1, 270. doi: 10.1136/ bmj.1.1466.270-c
Résumé PubMed / Texte intégral Croisé / Google Scholar
Il s’agit de l’un des plus importants ouvrages de l’histoire de l’art et de l’histoire de l’Art. Structure et propriétés de l’exopolysaccharide produit par Streptococcus macedonicus Sc136. Glycobiologie 11, 131-139. doi: 10.1093/glycob|11.2.131
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Wang, L., Christophersen, C. T., Sorich, M. J., Gerber, J. P., Angley, M. T. et Conlon, M.A. (2013). Augmentation de l’abondance de Sutterella spp. et les couples de ruminococcus dans les selles d’enfants atteints de troubles du spectre autistique. Mol. Autisme 4, 42. doi: 10.1186/2040-2392-4-42
Résumé PubMed / Texte intégral Croisé / Google Scholar
Il s’agit de l’un des plus grands ouvrages de l’histoire de l’art et de l’histoire de l’Art. Métagénome du lait humain: une analyse de la capacité fonctionnelle. Microbiol BMC. 13:116. doi: 10.1186/1471-2180-13-116
Résumé PubMed / Texte intégral Croisé / Google Scholar
Watanabe, K., Fujimoto, J., Tomii, Y., Sasamoto, M., Makino, H., Kudo, Y., et al. (2009). Lactobacillus kisonensis sp. nov., Lactobacillus otakiensis sp. nov., Lactobacillus rapi sp. nov. et Lactobacillus sunkii sp. nov., espèce hétérofermentative isolée du sunki, un cornichon japonais traditionnel. Int. J. Syst. Evol. Microbiol. 59 (Pt 4), 754-760. doi: 10.1099/ijs.0.004689-0
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Wu, R., Wang, L., Wang, J., Li, H., Menghe, B., Wu, J., et al. (2009). Isolement et sélection probiotique préliminaire de lactobacilles de koumiss en Mongolie intérieure. J. Microbiol basique. 49, 318–326. doi: 10.1002 / jobm.200800047
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Il s’agit de la première édition de la série. Velvet: algorithmes pour l’assemblage de lecture courte de novo utilisant des graphes de Bruijn. Genome Res. 18, 821-829. doi: 10.1101/gr.074492.107
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Zhang, W., Sun, Z., Sun, T., et Zhang, H. (2010a). Criblage par PCR et analyse de séquences de clusters iol chez des souches de Lactobacillus casei isolées de koumiss. Folia Microbiol. (Praha) 55, 603-606. doi: 10.1007 / l12223-010-0097-3
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Zhang, W., Yu, D., Sun, Z., Wu, R., Chen, X., Chen, W., et al. (2010b). Séquence complète du génome de Lactobacillus casei Zhang, une nouvelle souche probiotique isolée du koumiss traditionnel fait maison en Mongolie intérieure, en Chine. J. Bacteriol. 192, 5268–5269. doi: 10.1128/JB.00802-10
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Zhang, W., et Zhang, H. (2011). « Fermentation et koumiss », dans Handbook of Food and Beverage Fermentation Technology, 2e Éd., éd. Y. H. Hui (Boca Raton, FL: CRC Press), 165-172.
Google Scholar
Zhang, W. Y., Chen, Y. F., Zhao, W. J., Kwok, L. Y. et Zhang, H. P. (2015). Expression génique du système protéolytique de Lactobacillus helveticus H9 pendant la fermentation du lait. Ann. Microbiol. 65, 1171–1175. doi: 10.3168/jds.2014-8520
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Zhao, W., Chen, Y., Sun, Z., Wang, J., Zhou, Z., Sun, T., et al. (2011). Séquence génomique complète de Lactobacillus helveticus H10. J. Bacteriol. 193, 2666–2667. doi: 10.1128/JB.00166-11
Résumé PubMed / Texte intégral croisé / Google Scholar