- Introduction
- materialen en methoden
- bereiding van monsters
- Gradiëntverdunning en multiple Displacement amplificatie
- Bibliotheekconstructie en Sequencing
- gegevensanalyse
- controle van de Sequentiekwaliteit en Filtering
- Taxonomische toewijzing en diversiteit
- lezen assemblage, Genvoorspelling en annotatie
- nucleotidesequentie Toetredingsnummers
- resultaten
- experimentele opzet en Sequencing
- Taxonomische annotatie
- Metagenomische assemblage, Genvoorspelling en functionele annotatie
- discussie
- conclusie
- Auteursbijdragen
- belangenverstrengeling verklaring
- erkenning
- aanvullend materiaal
Introduction
Koumiss, ook chige, Chigo, arrag of airag genoemd, is een traditioneel gefermenteerd zuivelproduct. Het is al eeuwenlang een populair voedsel in Mongolië en binnen-Mongolië van China (Zhang and Zhang, 2011). Mensen in deze regio ‘ s gebruikten koumiss tijdens grote festiviteiten en offerplechtigheden (Zhang en Zhang, 2011). De vroegste vermelding van de productie van koumiss kan worden teruggevoerd tot de Han-dynastie (BC202-AD202). Dit product had bereikt wijdverspreide populariteit tijdens de Yuan-dynastie (AD1271-AD1368) (Zhang et al., 2010b). Tegenwoordig is koumiss een veel voorkomend voedsel voor de lokale bevolking van Mongolië en binnen-Mongolië, hoewel het slechts in enkele van deze gebieden op industriële schaal wordt geproduceerd. Koumiss biedt niet alleen rijke voedingsstoffen, waaronder een hoog gehalte aan essentiële aminozuren en vitamines, maar wordt ook verondersteld om een breed scala van medische aandoeningen te verlichten en is gunstig voor postoperatieve zorg (Jagielski, 1877; Thompson, 1879).
koumiss wordt traditioneel geproduceerd in houten vaten, containers gemaakt van dierenhuid of urnen. Fermentatie vindt natuurlijk plaats bij omgevingstemperatuur na toevoeging van gefiltreerde merriemelk in de container met oude koumiss, die dient als startcultuur (Zhang and Zhang, 2011). Koumiss is een goede bron van nieuwe bacteriën van biotechnologie potentieel (Zhang et al., 2010a; Pan et al., 2011). Daarom is het van groot belang om zoveel mogelijk met fermentatie geassocieerde koumissbacteriën te onderzoeken en te behouden. Tijdens de laatste decennia werden een aantal studies uitgevoerd om de koumiss bacteriële gemeenschap te onderzoeken (Wu et al., 2009; Hao et al., 2010), voornamelijk bestudeerd door cultuur -, moleculaire biologie-en pyrosequencing-gebaseerde methoden (Sun et al., 2010). Onder deze verschillende benaderingen, heeft de op pyrosequencing-gebaseerde methode het meest uitgebreide microbiota-profiel van koumiss onafhankelijk van fenotypic eigenschappen en problemen van cultivability van de individuele microben verstrekt. Echter, het spectrum van functionele genen gecodeerd door de koumiss bacteriën en hun fermentatieve capaciteiten blijven slecht gekenmerkt, in het bijzonder voor de zeldzame microbiële populaties.
in deze studie werd gebruik gemaakt van de eencellige genomica-techniek om de bacteriële metagenomen te analyseren van 10 koumissmonsters uit Mongolië en binnen-Mongolië in China. Het huidige werk heeft state-of-art technologieën toegepast in het onderzoeken van de bacteriële diversiteit in zuivelproducten. Ons werk heeft de haalbaarheid aangetoond van het ontdekken van laag-overvloedige taxa door de eencellige metagenomics benadering toe te passen. De bemoedigende resultaten zouden de ontwikkeling en toepassing van nieuwe benaderingen bij de aanpak van problemen in een traditioneel onderzoeksgebied bevorderen.
materialen en methoden
bereiding van monsters
in totaal werden 10 koumissmonsters verzameld uit Mongolië (MG14, MG15, MG16, MG17 en MG18) en binnen-Mongolië van China (NM17, NM18, NM19, NM20 en NM21) voor het metagenomics-onderzoek. Monsters werden aseptisch verzameld en vervoerd in droogijs.
een milliliter van elk monster werd voorbehandeld volgens de methode beschreven in Ward et al. (2013) met enkele wijzigingen. Kort, werden de monsters ontdooid in een ijsbad gedurende 3-5 minuten. Nadat de steekproeven gesmolten waren, werden zij onderworpen aan lage snelheids centrifugatie om onzuiverheden en eukaryotic celklonten te verwijderen. Vervolgens werden prokaryotische cellen uit de melksera gepelleteerd door middel van centrifugering bij 13.000 × g gedurende 15 minuten. De pellets werden opnieuw gesuspendeerd in 2 mL fosfaatgebufferde zoutoplossing (PBS) met 1% Triton X-100 en geïncubeerd gedurende 2 uur bij 37°C om eventuele resterende eukaryotische cellen te verwijderen. Vervolgens werden bacteriën gepelleteerd door centrifugeren bij 13.000 × g gedurende 15 minuten en de pellets werden opnieuw gesuspendeerd in 500 µL PBS. Tenslotte werd de centrifugatiestap nogmaals herhaald om de bacteriële cellen te wassen.
Gradiëntverdunning en multiple Displacement amplificatie
om de bacteriën met een gering aantal bacteriën te detecteren, werd de bacteriesuspensie van elk koumissmonster seriematig verdund voor een volgende amplificatiereactie. Het celaantal in elke steekproef werd ruwweg geschat onder een microscoop (Nikon, Tokyo, Japan) gebruikend een cel het tellen kamer (Qiujing, Shanghai, China). De verdunningsstap werd voortgezet tot het aantal cellen in elke bacteriesuspensie ongeveer 100 bedroeg. Meervoudige verdringingsversterking van de verdunde cellen werd uitgevoerd met behulp van de Repli-g eencellige Kit (Qiagen, Germantown, MD, USA) volgens de instructies van de fabrikant.
Bibliotheekconstructie en Sequencing
geamplificeerd DNA werd willekeurig geschoren en de fragmenten van ongeveer 500 bp werden geselecteerd. Na de bouw van de bibliotheek werden PerkinElmer LabChip ® GX Touch en StepOnePlusTM Real-Time PCR-systeem gebruikt voor de kwaliteitscontrole van de bibliotheek. Tot slot werden 125-bp gepaarde-end reads gesequenced op het Illumina hiseq 2500 platform volgens de instructies van de fabrikant.
gegevensanalyse
controle van de Sequentiekwaliteit en Filtering
ruwe reads die door de sequencer worden gegenereerd, kunnen kunstmatige reads bevatten van adapterverontreiniging tijdens de bouw van de bibliotheek. Daarom werden drie stappen uitgevoerd om een hoogwaardige schone leesdataset te verkrijgen: (1) eliminatie van reads veroorzaakt door adapterverontreiniging; (2) verwijdering van reads met een gemiddelde score onder een phred score van Q30, die werd beschouwd als de laagste cut-off voor een hoogwaardige base; (3) verwijdering van reads met een significante overmaat van “N” (≥5% van de read). De downstream-analyse was gebaseerd op de schone gegevens. Bovendien werd de statistische basiskwaliteit, gebaseerd op Q30 en de GC-inhoud, berekend.
uitlijningen tegen het gastheergenoom werden uitgevoerd om de gastheergebaseerde contaminantsequenties te verwijderen. Om het even welke gastheer-veroorzaakte leest werden weggegooid vóór verdere vergelijking met de verwijzingssequenties van het bacteriën (of virussen) genoom. Om nauwkeurigere resultaten te verkrijgen, werd het Burrows–Wheeler aligner (BWA) (versie 0.97 a) MEM model gebruikt in de alignments (Li and Durbin, 2009).
Taxonomische toewijzing en diversiteit
de websoftware Metaphlan werd gebruikt voor taxonomische toewijzing aan genus-en soortenniveaus (Segata et al., 2012). Om de diversiteit van soorten binnen en tussen monsters te vergelijken, analyseerden we de alfa – en bèta-diversiteit aan de hand van het R-gerelateerde pakket.
lezen assemblage, Genvoorspelling en annotatie
om meer uitgebreide informatie te verkrijgen, hebben we de geschoren fragmenten tot genoom (contigs) samengesteld. Nochtans, wegens de aanwezigheid van veelvoudige species, die in metagenomic steekproeven gemeenschappelijk is, verbeterden wij de methode van het het genoom assembleren van de bioinformatica die normaal voor enige speciesanalyse wordt gebruikt door SPAdes (versie 3.6.2), interne scripts, en metagenomic databases te integreren (Zerbino en Birney, 2008; Nurk et al., 2013).
de metagenemark software werd gebruikt voor genvoorspelling van de geassembleerde contigs (Noguchi et al., 2006). Redundante genen werden verwijderd met behulp van CD-HIT met de dekking van 90 en 95% identiteit (Li en Godzik, 2006; Fu et al., 2012). De relatieve abundanties van de genen werden bepaald door het rangschikken van Leest van uitstekende kwaliteit aan de gencatalogus gebruikend dezelfde procedure uit te lijnen. De downstream discrepant analyse was gebaseerd op de Gen relatieve abundanties. Genannotatie werd uitgevoerd door de sequenties van hoge kwaliteit uit te lijnen tegen verschillende openbare databases (namelijk NCBI non-redundant database, NR; Clusters van Orthologe groepen van eiwitten, radertjes; Kyoto Encyclopedie van genen en genomen, KEGG) met behulp van BLAST (Altschul et al., 1997). Een domein zoekopdracht werd uitgevoerd met behulp van Interproscan (Mulder en Apweiler, 2008).
nucleotidesequentie Toetredingsnummers
de sequentiegegevens die in deze studie zijn gerapporteerd, zijn gedeponeerd in de SRA-database (Toetredingsnr.SRP083102).
resultaten
experimentele opzet en Sequencing
om de weinig voorkomende bacteriën in koumiss op te sporen, werd de eencellige amplificatietechniek gebruikt om de metagenomen van de monsters te analyseren. Drie bacteriesuspensies, elk van ongeveer 100 cellen, werden afgeleid uit een onafhankelijk koumissmonster door seriële verdunning. In totaal werden 30 verdunde suspensies geanalyseerd. Aan elk verdund monster werd een andere monstercode gegeven, d.w.z. het identificatienummer van het monster gevolgd door 1, 2 of 3, die de drie afzonderlijke verdunningen vertegenwoordigt. Gebaseerd op een aanname dat sommige zeldzame soorten aanwezig zullen zijn in een van de verdunningen, werd meervoudige verplaatsingsamplificatie van de cellen uitgevoerd; en ongeveer 5 Gb gegevens werden gegenereerd voor elke koumiss bacteriële suspensie.
uit de 10 koumissmonsters werden in totaal 1.040.323.864 ruwe reads verkregen (in totaal 30 bacteriesuspensies). Het gemiddelde aantal reads voor elke 100-cel-suspensie was 34.677.462 (aanvullende tabel S1). Na het trimmen en filteren van de ongekwalificeerde sequenties, verkregen we 1.018.381.702 clean reads voor alle monsters (aanvullende tabel S1). De waarden van Shannon-index, Simpson-index, Chao1-index en het aantal waargenomen soorten (figuren 1-4) toonden aan dat de meeste koumiss-monsters een hoge bacteriële biodiversiteit hadden. De Shannon-Wiener diversiteitscurves toonden aan dat de sequentiediepte voldoende was voor alle monsters (figuur 1).
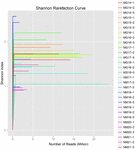
FIGUUR 1. Shannon rarefaction krommen die de microbiële diversiteit van koumiss steekproeven schatten.
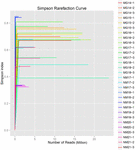
FIGUUR 2. Simpson rarefaction curves die de microbiële diversiteit van koumiss monsters schatten.
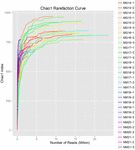
FIGUUR 3. Chao1 rarefaction krommen die de microbiële diversiteit van koumiss monsters schatten.
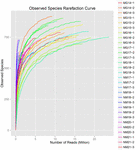
FIGUUR 4. Waargenomen species rarefaction index die de microbiële diversiteit van koumiss monsters schatten.
Taxonomische annotatie
de sequenties van hoge kwaliteit werden toegewezen aan verschillende taxonomische niveaus om een diepgaande analyse van de bacteriële gemeenschappen van het monster mogelijk te maken. Met verwijzing naar enkele gepubliceerde studies over de biodiversiteit van koumiss (Wu et al., 2009; Hao et al., 2010; Sun et al., 2010), classificeerden we de bekende en niet eerder gemelde koumiss-geassocieerde bacteriën als gemeenschappelijke en zeldzame taxa, respectievelijk.
de sequenties van hoge kwaliteit vertegenwoordigden 13 verschillende geslachten (Figuur 5). Drie van hen hadden een gemiddelde relatieve abundantie van meer dan 1%, met inbegrip van Lactobacillus (L.), Lactococcus, en Streptococcus. Vooral Lactobacillus en Lactococcus waren de twee meest voorkomende geslachten gevonden in de koumiss. Het aandeel Lactobacillus in de monsters varieerde van 52,72 tot 99,96%. Twee leden van dit geslacht, L. helveticus en L. kefiranofaciens, werden gedomineerd onder de meeste koumissmonsters (Figuur 6). De soort L. buchneri was vooral aanwezig in de Mongoolse monsters, terwijl Lactococcus lactis in de meeste monsters werd gedetecteerd, ongeacht het bemonsteringsgebied.
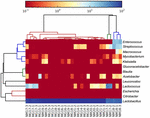
FIGUUR 5. Heatmap die de relatieve abundantie van bacteriën toont die in de koumissmonsters op genusniveau worden gedetecteerd.
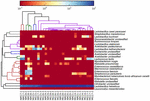
FIGUUR 6. Heatmap toont de relatieve abundantie van bacteriën gedetecteerd in de koumiss monsters op soortniveau.
Metagenomische assemblage, Genvoorspelling en functionele annotatie
het samenstellen van de reads resulteerde in een assemblagelengte van 614.392.623 bp. De N50-waarden voor de samenstellingen varieerden van 5,596 tot 35.200 bp (aanvullende tabel S2). Het aantal voorspelde genen in de koumiss-monsters varieerde van 10.347 tot 34.547, met een gemiddelde lengte variërend van 647.82 tot 985.33 bp (aanvullende tabel S3). Hoewel empirische gen-functionele analyses buiten het bereik van de huidige studie vielen, annoteerden we de bacteriële microbiomen van koumiss met behulp van de COG-en KEGG-databases, die de genfunctie voorspelden grotendeels gebaseerd op sequentiehomologie.
een totaal van 545 overeenkomsten in de annotatie-output toonde een hoge homologie aan de lactose-gebruiksgenen (COGs-categorie koolhydraten en metabolisme, G), en sommige van hen bevonden zich binnen hetzelfde contig (Tabel 1).
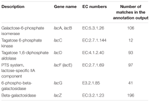
TABEL 1. Lactose metabolisme-gerelateerde genen geannoteerd in de koumiss bacteriële metagenome.
sommige andere sequenties kunnen coderen voor vermeende genen binnen de COGs-categorie aminozuurtransport en-metabolisme (E), waaronder sequenties die overeenkwamen met caseïneafbrekende proteïnasen, het Opp-systeem voor de opname van oligopeptiden van 4-18 residuen en aminopeptidasen (bijv. leucylaminopeptidase, peptidyldipeptidase A, aminopeptidase N, proline iminopeptidase en endopeptidase). Hoewel sommige sequenties grote overeenkomsten hadden met de aminotransferasen specifiek voor arginine, aspartaat, methionine en isoleucine, werd slechts één significante overeenkomst geïdentificeerd, die overeenkwam met een vermoedelijk van S. macedonicus afkomstig klasse I/klasse II domein (IPR004839)-bevattende aminotransferase specifiek voor tyrosine en fenylalanine. Tot slot deelden een aantal sequenties een hoge homologie aan de aminozuurlyasen, waaronder s-ribosylhomocysteïnelyase, argininosuccinaatlyase, aspartaat-ammoniak-lyase, cystathionine-gamma-lyase, histidine-ammoniak-lyase en O-acetylhomoserine (thiol) – lyase.
discussie
Koumiss is een populair traditioneel gefermenteerd zuivelproduct in Mongolië en binnen-Mongolië in China. Hoewel een aantal studies zijn uitgevoerd om de bacteriële diversiteit in koumiss te onderzoeken, is weinig informatie verkregen met betrekking tot de genetische capaciteit van de koumiss microbiota. Hier, pasten wij de eencellige amplificatietechniek toe om de bacteriële metagenomes van koumiss te analyseren, in het bijzonder die zich op de laag-overvloedige bacteriële bevolking concentreren.
de typische metagenomics benadering is eerder toegepast om de koumiss microbiota te beschrijven. Nochtans, wegens de hoge kosten om een diepe opeenvolging te bereiken, wordt de zeldzame microbiota bevolking in de steekproeven vaak ontoereikend behandeld. Aldus, blijven zowel phylogenic als functionele metagenomes van de minderheidsbacteriën beperkt. Onze eencellige amplificatiemethode omvatte de seriële verdunning van monsters tot suspensies van 100 cellen. Wegens het lage aantal cellen huidig in de verdunde koumiss steekproeven, zou slechts een beperkte hoeveelheid DNA worden geëxtraheerd. De versterkingsstap verhoogde de hoeveelheid te analyseren materialen van DNA en vergemakkelijkte vandaar de metagenome analyse van steekproeven die minieme hoeveelheden van DNA bevatten. Een nadeel van deze methode was de moeilijkheid om ervoor te zorgen dat sequenties van elk taxon in het proces even zouden worden versterkt; daarom konden de hier verkregen resultaten Alleen de relatieve abundanties van sequenties maar niet absolute hoeveelheden geïdentificeerde taxa of functionele genen weergeven. Toch zou dit weinig effect hebben op onze Analyse omdat de huidige studie verschilt van andere gepubliceerde werken in het focussen van de zeldzame bacteriële populatie. De door dit werk gegenereerde gegevens vormen een aanvulling op de ondervertegenwoordigde bevolking. Wij geloven dat de huidige aanpak geschikt is voor toekomstige analyse van bacteriediversiteit voor verschillende soorten ecologische omgevingen.
de koumiss bacteriële microbiota bestaat voornamelijk uit melkzuurbacteriën (LAB) en enkele azijnzuurbacteriën (Zhang and Zhang, 2011). Zoals verwacht, bevatte onze dataset voornamelijk sequenties die het LAB en azijnzuurbacteriën vertegenwoordigen. Sequenties die overeenkomen met Lactobacillus helveticus domineerden over alle monsters. Bovendien, sequenties die de soort, L. kefiranofaciens. L. buchneri. L. kefiranofaciens. Enterococcus (E.) casseliflavus. E. faecalis. E. faecium. Leuconostoc mesenteroides. Lactococcus lactis, en Acetobacter pasteurianus, werden ook gevonden. De identificatie van sequenties van verschillende taxa kan niet genoeg zijn om de levensvatbaarheid van de bacteriën aan te tonen, het weerspiegelt niettemin de bacteriële gemeenschap op een bepaald punt van het fermentatieproces. Miyamoto et al. (2010) suggereert dat de bacteriële prevalentie in de uiteindelijke gefermenteerde producten gerelateerd is aan hun zuurstress tolerantie. Over het algemeen hebben lactobacilli een hogere zuurtolerantie dan lactokokken, wat de hoge relatieve overvloed van lactobacilli-sequenties in onze dataset kan verklaren. Aan de andere kant, het frequente voorkomen van L. helveticus in koumiss viel samen met de waarneming van de dominantie van L. helveticus sequenties.
in onze dataset kwamen sommige sequenties overeen met L. otakiensis, een zeldzame soort die nooit is gerapporteerd in koumiss of andere zuivelniches. De soort werd eerst beschreven en geïsoleerd uit de niet-gezouten beitsoplossing die wordt gebruikt bij de productie van sunki, een traditionele Japanse augurk (Watanabe et al., 2009). Het werd ontdekt door het versterkte polymorfisme van de fragmentlengte dat op het recA-gen wordt gebaseerd profileren. Sindsdien is deze soort niet geassocieerd met andere voedselgerelateerde niches. Zo is het waarschijnlijk dat deze bacterie behoort tot de autochtone flora van augurken. We kunnen echter niet uitsluiten dat het niet is gevonden alleen vanwege de gevoeligheid van de gebruikte detectiemethode. Lactobacillus otakiensis kan d-vertakte aminozuren produceren, zoals d-leucine, D-allo-isoleucine en d-valine (Doi et al., 2013). Het kan worden gebruikt om de productiekenmerken van bepaalde gefermenteerde levensmiddelen te verbeteren (Kato et al., 2011).
onze studie identificeerde ook sequenties die de soort S. macedonicus vertegenwoordigen, die nooit is gerapporteerd als een koumiss-geassocieerde bacterie. In plaats daarvan, het is een starter cultuur aanwezig in de Griekse schapen-en geitenkaas (Georgalaki et al., 2000). Sommige leden van deze soort zijn in staat om exopolysacchariden, bacteriocinen (Vincent et al., 2001; Anastasiou et al., 2015), en gamma-aminoboterzuur (Franciosi et al., 2015). Hoewel deze soort vaak wordt geïsoleerd uit gefermenteerd voedsel, is de oorspronkelijke niche van S. macedonicus controversieel geweest (Guarcello et al., 2016). Niet tot voor kort, Papadimitriou et al. (2015) identificeerde een verworven plasmide, pSMA198, van Lactococcus lactis. Het plasmide werd waarschijnlijk overgedragen via een voorouderlijke genetische uitwisseling gebeurtenis binnen een zuivelproductomgeving, zinspelend op de zuivel oorsprong van S. macedonicus (Papadimitriou et al., 2015). Vergelijkbaar met S. thermophilus. S. macedonicus is nauw verwant aan de commensalen en opportunistische pathogenen van het S. bovis/S. equinus complex.
de identificatie van sequenties van de soort ruminococcus torques was onverwacht, aangezien deze bacterie gewoonlijk in de darmomgeving wordt aangetroffen. Het is een normale menselijke darm microbe die mucine oligosachariden kan degraderen door constitutieve productie van secretoire glycosidasen (Hoskins et al., 1985). Recent klinisch bewijs toont aan dat de fecale overvloed van deze soort is veranderd in kinderen met autisme spectrum stoornis; echter, zijn rol in de aandoening blijft onduidelijk (Wang et al., 2013).
verrassend genoeg vertegenwoordigden sommige sequenties potentiële pathogenen. Bijvoorbeeld, Klebsiella longontsteking is een opportunistische menselijke pathogeen die in ongeveer 40% van de menselijke en dierlijke darmen woont. Mycobacterium orygis, voorheen de Oryx bacillus genoemd, is een lid van het Mycobacterium tuberculosis complex dat menselijke tuberculose kan veroorzaken (Dawson et al., 2012). Deze twee soorten zijn gemeld in rauwe melk maar niet in koumiss. Daarom kan hun aanwezigheid toe te schrijven aan de verontreiniging tijdens conventionele koumiss productie, met name onder niet – of lage aseptische manipulatie omstandigheden.
bacterieel metabolisme speelt een belangrijke rol bij de vorming van de eigenschappen en kwaliteit van koumiss. Microbiële processen zoals lipolyse en proteolyse zijn nodig voor het synthetiseren van koumiss aromatische en smaakstoffen (Gesudu et al., 2016). Consequent, bevatte het bacteriële metagenome opeenvolgingen die potentieel voor degradatie van lactose en proteolytic systemen coderen. In tegenstelling tot de relatief eenvoudige lactose katabole wegen, De Lab proteolytische systemen zijn opgebouwd uit een diverse reeks van enzymen (Chen et al., 2014). Vergeleken met andere gedetecteerde koumiss LAB, wordt de dominante soort L. helveticus gekenmerkt met een hoge proteolytische activiteit (Zhang et al., 2015). Het meeste laboratorium bezit slechts één cel-envelop proteïnase dat de melk caseïne hydrolyse in werking stelt, terwijl L. helveticus minstens twee van deze enzymen, namelijk PrtH en PrtH2 (Zhao et al., 2011). Aldus, kunnen de hoge verhoudingen van opeenvolgingen die aan de sterke proteolytic species van L. helveticus en proteolysis-verwante genen corresponderen met de vrij hoge inhoud van peptides en vrije aminozuren in koumiss.
een probleem bij de productie van koumiss op industriële schaal is de controle van de smaakperceptie, omdat koumiss traditioneel wordt gemaakt door natuurlijke fermentatie. Zo is het moeilijk geweest om de smaak en het profiel van de belangrijkste smaakcomponenten te definiëren, met name in de aanwezigheid van de natuurlijke verontreinigingen. De productie van belangrijke smaakcomponenten is het resultaat van fermentatieve en enzymatische afbraak van aminozuren, zoals de vertakte aminozuren, methionine en aromatische aminozuren (Ardo, 2006). Voorbeelden van smaakcomponenten zijn Aldehyden, organische zuren en esters, die worden gevormd door transaminase (AT)-Route (Helinck et al., 2004). Deze route wordt geïnitieerd door een transaminase die de omzetting van een aminozuur in het overeenkomstige α-ketozuur katalyseert (Helinck et al., 2004). Sommige sequenties in onze dataset kwamen overeen met vermeende vertakte keten en aromatische aminozuuraminotransferases. In het bijzonder vonden we een vermoedelijk aromatisch aminozuur aminotransferase I in het contig van de koumiss-geassocieerde soort, S. macedonicus. Aangezien ons huidige werk slechts microbiome in silico annoteerde, zijn onze gegevens niet genoeg om aan te tonen dat deze geà dentificeerde gensequenties inderdaad functioneel waren om de bovengenoemde koumissaromasamenstellingen te produceren. De aanwezigheid van deze genen stelt niettemin voor dat zij sommige mogelijke kandidaten voor dergelijke fermentatieve activiteiten zijn; nochtans, zou het verdere experimentele werk worden vereist om hun nauwkeurige functionele rollen te verduidelijken.
bovendien vonden we sequenties die overeenkomen met andere aminozuuromzettingsroutes, waaronder aminozuurlyasen en threonine-aldolase. Het voormalige enzym katalyseert de omzetting van methionine in methaanthiol (Irmler et al., 2008), wat resulteert in de vorming van dimethyldisulfide en dimethyltrisulfide (Fernandez et al., 2000), terwijl de laatste de omzetting van threonine in glycine en aceetaldehyde katalyseert (Ott et al., 2000). Evenzo, kan het slechts lokaliseren van deze opeenvolgingen binnen koumiss microbiome niet als definitief bewijsmateriaal in hun daadwerkelijke biologische rollen dienen; de toekomstige experimentele bevestiging zal worden vereist.
conclusie
deze studie maakte gebruik van een gemodificeerde metagenomicsmethode om het bacteriële microbioom van koumissmonsters uit Mongolië en binnen-Mongolië van China te analyseren. Wij kenmerkten zowel de phylogenic als functionele metagenomes van de zeldzame species in koumiss; en onze dataset weerspiegelt eigenschappen die van biotechnologiebelang en potentieel zijn. Onze studie heeft voor het eerst de haalbaarheid aangetoond van het opnemen van de eencellige amplificatietechnieken in het ontdekken van koumiss bacteriële microbiota, evenals microbiële contaminanten. De hierin ontwikkelde technieken kunnen in toekomstige studies worden gebruikt om veranderingen in koumiss microbiome langs het fermentatieproces te controleren, met nadruk op de microbiële minderheid bevolking. Voorts kunnen andere omicstechnieken, zoals transcriptomics, metabolomics, aan de huidige metagenomicsanalyse worden gekoppeld om de functies en metabolische capaciteit van koumiss microbiome te bevestigen.
technisch gezien zou de volgende stap van dit werk het optimaliseren van de huidige methode zijn. Bijvoorbeeld, door de steekproefverdunning vóór de versterking van DNA te verhogen, kan de kans om zeldzame en nieuwe bacteriën aan het licht te brengen worden verbeterd. Anderzijds, kan een alternatieve het rangschikken techniek die lange leest, zoals vreedzame Biosciences enige molecule, real-time het rangschikken technologie kan produceren, worden aangewend om het genoom het assembleren proces te verbeteren.
Auteursbijdragen
WZ en HZ ontwierpen de studie. WZ, GY, JY en L-YK schreven het manuscript. QH, WH, WL, BM en TS voerden experimenten uit. WZ en QH analyseerden gegevens. Alle auteurs bekeken het manuscript.
belangenverstrengeling verklaring
de auteurs verklaren dat het onderzoek werd uitgevoerd zonder enige commerciële of financiële relatie die als een potentieel belangenconflict kon worden opgevat.
erkenning
dit onderzoek werd ondersteund door de National Natural Science Foundation of China (subsidie nr. 31571815).
aanvullend materiaal
het aanvullende materiaal voor dit artikel is online te vinden op: https://www.frontiersin.org/article/10.3389/fmicb.2017.00165/full#supplementary-material
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST en PSI-BLAST: een nieuwe generatie van eiwit database zoekprogramma ‘ s. Nucleïnezuren Res. 25, 3389-3402. doi: 10.1093/nar / 25.17.3389
PubMed Abstract / CrossRef Full Text / Google Scholar
Anastasiou, R., Driessche, G. V., Boutou, E., Kazou, M., Alexandraki, V., Vorgias, C. E., et al. (2015). Geconstrueerde stammen van Streptococcus macedonicus naar een osmotisch stress resistent fenotype behouden hun vermogen om de bacteriocin macedocin onder hyperosmotische omstandigheden te produceren. J. Biotechnol. 212, 125–133. doi: 10.1016/j.jbiotec.2015.08.018
PubMed Abstract / CrossRef Full Text / Google Scholar
Ardo, Y. (2006). Smaakvorming door aminozuurkatabolisme. Biotechnol. ADV. 24, 238-242. doi: 10.1016 / j.biotechadv.2005.11.005
PubMed Abstract / CrossRef Full Text / Google Scholar
Chen, Y. F., Zhao, W. J., Wu, R. N., Sun, Z. H., Zhang, W. Y., Wang, J. C., et al. (2014). Proteoomanalyse van Lactobacillus helveticus H9 tijdens groei in magere melk. J. Dairy Sci. 97, 7413–7425. doi: 10.3168 / jds.2014-8520
PubMed Abstract / CrossRef Full Text / Google Scholar
Dawson, K. L., Bell, A., Kawakami, R. P., Coley, K., Yates, G., and Collins, D. M. (2012). Overdracht van Mycobacterium orygis (M. tuberculosis complex species) van een tuberculose-patiënt naar een melkkoe in Nieuw-Zeeland. J. Clin. Microbiol. 50, 3136–3138. doi: 10.1128 / JCM.01652-12
PubMed Abstract / CrossRef Full Text / Google Scholar
Doi, K., Mori, K., Mutauchi, Y., Tashiro, K., Fujino, Y., Ohmori, T., et al. (2013). Concept genoomsequentie van D-vertakte keten aminozuur producent Lactobacillus otakiensis JCM 15040T, geïsoleerd uit een traditionele Japanse augurk. Genoom Kondigt Aan. 1, e546-e513. doi: 10.1128 / genomeA.00546-13
PubMed Abstract / CrossRef Full Text / Google Scholar
Fernandez, M., Van Doesburg, W., Rutten, G. A., Marugg, J. D., Alting, A. C., Van Kranenburg, R., et al. (2000). Moleculaire en functionele analyses van het METC-gen van Lactococcus lactis, dat codeert voor cystathionine-bèta-lyase. Appl. Environ. Microbiol. 66, 42–48. doi: 10.1128 / AEM.66.1.42-48.2000
PubMed Abstract / CrossRef Full Text / Google Scholar
Franciosi, E., Carafa, I., Nardin, T., Schiavon, S., Poznanski, E., Cavazza, A., et al. (2015). Biodiversiteit en gamma-aminoboterzuur productie door melkzuurbacteriën geïsoleerd uit traditionele alpine rauwe koemelk kazen. Biomed Res. Int. 2015, 625740. doi: 10.1155/2015/625740
PubMed Abstract / CrossRef Full Text / Google Scholar
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: versneld voor het clusteren van de volgende generatie sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
PubMed Abstract | CrossRef Full Text | Google Scholar
Georgalaki, M. D., Sarantinopoulos, P., Ferreira, E. S., De Vuyst, L., Kalantzopoulos, G., and Tsakalidou, E. (2000). Biochemical properties of Streptococcus macedonicus strains isolated from Greek Kasseri cheese. J. Appl. Microbiol. 88, 817–825. doi: 10.1046/j.1365-2672.2000.01055.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Gesudu, Q. M., Zheng, Y., Xi, X. X., Hou, Q. C., Xie, H. Y., Huang, W. Q., et al. (2016). Het onderzoeken van bacteriële populatiestructuur en dynamica in traditionele koumiss van binnen Mongolië gebruikend enig molecuul real-time het rangschikken. J. Dairy Sci. 99, 7852–7863. doi: 10.3168 / jds.2016-11167
PubMed Abstract / CrossRef Full Text / Google Scholar
Guarcello, R., Carpino, S., Gaglio, R., Pino, A., Rapisarda, T., Caggia, C., et al. (2016). Een grote fabriek-schaal toepassing van geselecteerde autochtone melkzuurbacteriën voor de productie van PDO Pecorino Siciliano kaas. Voedsel Microbiol. 59, 66–75. doi: 10.1016 / j. fm.2016.05.011
PubMed Abstract / CrossRef Full Text / Google Scholar
Hao, Y., Zhao, L., Zhang, H., Zhai, Z., Huang, Y., Liu, X., et al. (2010). Identificatie van de bacteriële biodiversiteit in koumiss door denaturering van gradiëntgelelektroforese en soortspecifieke polymerasekettingreactie. J. Dairy Sci. 93, 1926–1933. doi: 10.3168 / jds.2009-2822
PubMed Abstract / CrossRef Full Text / Google Scholar
Helinck, S., Le Bars, D., Moreau, D., and Yvon, M. (2004). Vermogen van thermofiele melkzuurbacteriën om aromaverbindingen van aminozuren te produceren. Appl. Environ. Microbiol. 70, 3855–3861. doi: 10.1128 / AEM.70.7.3855-3861.2004
PubMed Abstract / CrossRef Full Text / Google Scholar
Hoskins, L. C., Agustines, M., McKee, W. B., Boulding, E. T., Kriaris, M., and Niedermeyer, G. (1985). Mucine afbraak in menselijke dikke darm ecosystemen. Isolatie en eigenschappen van fecale stammen die ABH bloedgroep antigenen en oligosacchariden afbreken van mucine glycoproteïnen. J. Clin. Investeren. 75, 944–953. doi: 10.1172 / JCI111795
PubMed Abstract / CrossRef Full Text / Google Scholar
Irmler, S., Raboud, S., Beisert, B., Rauhut, D., and Berthoud, H. (2008). Klonen en karakterisering van twee Lactobacillus casei genen die coderen voor een cystathionine lyase. Appl. Environ. Microbiol. 74, 99–106. doi: 10.1128 / AEM.00745-07
PubMed Abstract / CrossRef Full Text / Google Scholar
Jagielski, V. (1877). De waarde van koumiss bij de behandeling van misselijkheid, braken en onvermogen om ander voedsel op de maag te behouden. Br. Med. J. 2, 919-921. doi: 10.1136/bmj.2.887.919
PubMed Abstract / CrossRef Full Text / Google Scholar
Kato, S., Ishihara, T., Hemmi, H., Kobayashi, H., and Yoshimura, T. (2011). Veranderingen in D-aminozuurconcentraties en microbiële gemeenschapsstructuren tijdens de fermentatie van rode en witte wijn. J. Biosci. Bioeng. 111, 104–108. doi: 10.1016/j.jbiosc.2010.08.019
PubMed Abstract / CrossRef Full Text / Google Scholar
Li, H., and Durbin, R. (2009). Snelle en nauwkeurige korte uitlijning met Burrows-Wheeler transformator. Bioinformatica 25, 1754-1760. doi: 10.1093 / bioinformatics / btp324
PubMed Abstract / CrossRef Full Text / Google Scholar
Li, W., and Godzik, A. (2006). Cd-hit: een snel programma voor het clusteren en vergelijken van grote reeksen proteã ne of nucleotideopeenvolgingen. Bioinformatica 22, 1658-1659. doi: 10.1093 / bioinformatics / btl158
PubMed Abstract / CrossRef Full Text / Google Scholar
Miyamoto, M., Seto, T., Nakajima, H., Burenjargal, S., Gombojav, A., Demberei, S., et al. (2010). Denaturing gradient gel electroforese analyse van melkzuurbacteriën en gisten in traditionele Mongoolse gefermenteerde melk. Voedsel Sci. Technol. 16, 319-326. doi: 10.3136/fstr.16.319
CrossRef Full Text / Google Scholar
Mulder, N. J., and Apweiler, R. (2008). De InterPro database en hulpmiddelen voor eiwitdomeinanalyse. Curr. Protocol. Bioinformatica Hoofdstuk 2, Unit2.7. doi: 10.1002 / 0471250953. bi0207s21
PubMed Abstract / CrossRef Full Text / Google Scholar
Noguchi, H., Park, J., and Takagi, T. (2006). Metageen: prokaryotic gen het vinden van van het omgevingsgenoom shotgun opeenvolgingen. Nucleïnezuren Res. 34, 5623-5630. doi: 10.1093 / nar / gkl723
PubMed Abstract / CrossRef Full Text / Google Scholar
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A. A., Korobeynikov, A., Lapidus, A., et al. (2013). Het assembleren van eencellige genomen en mini-metagenomen uit chimerische MDA-producten. J. Comput. Biol. 20, 714–737. doi: 10.1089 / cmb.2013.0084
PubMed Abstract / CrossRef Full Text / Google Scholar
Ott, A., Germond, J. E., en Chaintreau, A. (2000). Oorsprong van aceetaldehyde tijdens de melkfermentatie met behulp van (13)C-gelabelde precursoren. J. Agric. Voedselchem. 48, 1512–1517. doi: 10.1021 / jf9904867
PubMed Abstract / CrossRef Full Text / Google Scholar
Pan, D. D., Zeng, X. Q., and Yan, Y. T. (2011). Karakterisering van Lactobacillus fermentum SM-7 geïsoleerd uit koumiss, een potentiële probiotische bacterie met cholesterolverlagende effecten. J. Sci. Voedsel. Agric. 91, 512–518. doi: 10.1002 / jsfa.4214
PubMed Abstract / CrossRef Full Text / Google Scholar
Papadimitriou, K., Anastasiou, R., Maistrou, E., Plakas, T., Papandreou, N. C., Hamodrakas, S. J., et al. (2015). Acquisitie door horizontale genoverdracht van plasmide pSMA198 door Streptococcus macedonicus ACA-DC 198 wijst naar de zuiveloorsprong van de soort. PLoS ONE 10: e0116337. doi: 10.1371 / journal.pone.0116337
PubMed Abstract / CrossRef Full Text / Google Scholar
Segata, N., Waldron, L., Ballarini, A., Narasimhan, V., Jousson, O., and Huttenhower, C. (2012). Het metagenomic microbiële gemeenschap profileren gebruikend unieke clade-specifieke teller genen. Nat. Methods 9, 811-814. doi: 10.1038 / nmeth.2066
PubMed Abstract / CrossRef Full Text / Google Scholar
Sun, Z., Liu, W., Zhang, J., Yu, J., Zhang, W., Cai, C., et al. (2010). Identificatie en karakterisering van de dominante lactobacillen geïsoleerd uit koumiss in China. J.-Generaal Appl. Microbiol. 56, 257–265. doi: 10.2323 / jgam.56.257
PubMed Abstract / CrossRef Full Text / Google Scholar
Thompson, J. (1879). De waarde van koumiss in het verspillen van ziekten. Br. Med. J. 1, 270. doi: 10.1136 / bmj.1.1466.270-c
PubMed Abstract / CrossRef Full Text / Google Scholar
Vincent, S. J., Faber, E. J., Neeser, J. R., Stingele, F., and Kamerling, J. P. (2001). Structuur en eigenschappen van het exopolysaccharide geproduceerd door Streptococcus macedonicus Sc136. Glycobiologie 11, 131-139. doi: 10.1093 / glycob / 11.2.131
PubMed Abstract / CrossRef Full Text / Google Scholar
Wang, L., Christophersen, C. T., Sorich, M. J., Gerber, J. P., Angley, M. T., and Conlon, M. A. (2013). Verhoogde abundantie van Sutterella spp. en ruminococcus torques in uitwerpselen van kinderen met autismespectrumstoornis. Mol. Autisme 4, 42. doi: 10.1186/2040-2392-4-42
PubMed Abstract / CrossRef Full Text / Google Scholar
Ward, T. L., Hosid, S., Ioshikhes, I., en Altosaar, I. (2013). Metagenome voor menselijke melk: een analyse van de functionele capaciteit. BMC Microbiol. 13:116. doi: 10.1186/1471-2180-13-116
PubMed Abstract / CrossRef Full Text / Google Scholar
Watanabe, K., Fujimoto, J., Tomii, Y., Sasamoto, M., Makino, H., Kudo, Y., et al. (2009). Lactobacillus kisonensis sp. nov., Lactobacillus otakiensis sp. nov., Lactobacillus rapi sp. nov. en Lactobacillus sunkii sp. nov. heterofermentatieve soorten geïsoleerd uit sunki, een traditionele Japanse augurk. Int. J. Syst. Evol. Microbiol. 59 (Pt 4), 754-760. doi: 10.1099 / ijs.0,004689-0
PubMed Abstract / CrossRef Full Text / Google Scholar
Wu, R., Wang, L., Wang, J., Li, H., Menghe, B., Wu, J., et al. (2009). Isolatie en voorlopige probiotische selectie van lactobacillen uit koumiss in binnen-Mongolië. J. Basic Microbiol. 49, 318–326. doi: 10.1002 / jobm.200800047
PubMed Abstract / CrossRef Full Text / Google Scholar
Zerbino, D. R., and Birney, E. (2008). Velvet: algoritmen voor de novo short read assembly met behulp van de Bruijn grafieken. Genome Res. 18, 821-829. doi: 10.1101 / gr.074492.107
PubMed Abstract / CrossRef Full Text / Google Scholar
Zhang, W., Sun, Z., Sun, T. en Zhang, H. (2010a). PCR screening en sequentie analyse van IOL clusters in Lactobacillus casei stammen geïsoleerd uit koumiss. Folia Microbiol. (Praha) 55, 603-606. doi: 10.1007 / s12223-010-0097-3
PubMed Abstract / CrossRef Full Text / Google Scholar
Zhang, W., Yu, D., Sun, Z., Wu, R., Chen, X., Chen, W., et al. (2010b). Volledige genoomsequentie van Lactobacillus casei Zhang, een nieuwe probiotische stam geïsoleerd uit traditionele zelfgemaakte koumiss in binnen-Mongolië, China. J. Bacteriol. 192, 5268–5269. doi: 10.1128 / JB.00802-10
PubMed Abstract / CrossRef Full Text / Google Scholar
Zhang, W., and Zhang, H. (2011). “Fermentation and koumiss,” in Handbook of Food and Beverage Fermentation Technology, 2nd Edn, ed. Y. H. Hui (Boca Raton, FL: CRC Press), 165-172.
Google Scholar
Zhang, W. Y., Chen, Y. F., Zhao, W. J., Kwok, L. Y., and Zhang, H. P. (2015). Genexpressie van proteolytisch systeem van Lactobacillus helveticus h9 tijdens melkfermentatie. Anne. Microbiol. 65, 1171–1175. doi: 10.3168 / jds.2014-8520
PubMed Abstract / CrossRef Full Text / Google Scholar
Zhao, W., Chen, Y., Sun, Z., Wang, J., Zhou, Z., Sun, T., et al. (2011). Volledige genoomsequentie van Lactobacillus helveticus H10. J. Bacteriol. 193, 2666–2667. doi: 10.1128 / JB.00166-11
PubMed Abstract / CrossRef Full Text / Google Scholar