- Einleitung
- Materialien und Methoden
- Probenvorbereitung
- Gradientenverdünnung und Multiple-Displacement-Amplifikation
- Bibliothekskonstruktion und Sequenzierung
- Datenanalyse
- Sequenzqualitätsprüfung und Filterung
- Taxonomische Zuordnung und Diversität
- Assemblierung, Genvorhersage und Annotation lesen
- Nucleotide Sequence Accession Numbers
- Ergebnisse
- Versuchsaufbau und Sequenzierung
- Taxonomische Annotation
- Metagenomische Assemblierung, Genvorhersage und funktionelle Annotation
- Schlussfolgerung
- Autorenbeiträge
- Interessenkonflikterklärung
- Anerkennung
- Ergänzungsmaterial
Einleitung
Koumiss, auch chige, chigo, arrag oder airag genannt, ist in der mongolischen Sprache eine Art traditionelles fermentiertes Milchprodukt. Es ist seit Jahrhunderten ein beliebtes Essen in der Mongolei und der Inneren Mongolei Chinas (Zhang und Zhang, 2011). Die Menschen in diesen Regionen konsumierten Koumiss während großer Feste und Opfergaben (Zhang und Zhang, 2011). Die früheste Aufzeichnung der Koumiss-Produktion geht auf die Han-Dynastie zurück (BC202-AD202). Dieses Produkt hatte während der Yuan-Dynastie (AD1271-AD1368) große Popularität erlangt (Zhang et al., 2010b). Heutzutage ist Koumiss ein verbreitetes Lebensmittel für die einheimische Bevölkerung der Mongolei und der Inneren Mongolei, obwohl es nur in wenigen dieser Gebiete im industriellen Maßstab hergestellt wird. Koumiss liefert nicht nur reichhaltige Nährstoffe, einschließlich eines hohen Gehalts an essentiellen Aminosäuren und Vitaminen, sondern lindert auch eine Vielzahl von Erkrankungen und ist für die postoperative Versorgung von Vorteil (Jagielski, 1877; Thompson, 1879).
Traditionell wird Koumiss üblicherweise in Holzfässern, Behältern aus Tierhaut oder Urnen hergestellt. Die Fermentation erfolgt auf natürliche Weise bei Raumtemperatur nach Zugabe von filtrierter Stutenmilch in den Behälter mit altem Koumiss, der als Starterkultur dient (Zhang und Zhang, 2011). Koumiss ist eine gute Quelle für neuartige Bakterien mit biotechnologischem Potenzial (Zhang et al., 2010a; Pan et al., 2011). Daher ist es von großem Interesse, möglichst viele fermentationsassoziierte Koumiss-Bakterien zu erforschen und zu konservieren. In den letzten Jahrzehnten wurden eine Reihe von Studien durchgeführt, um die Koumiss-Bakteriengemeinschaft zu untersuchen (Wu et al., 2009; Hao et al., 2010), hauptsächlich untersucht mit kultur-, molekularbiologischen und pyrosequenzierungsbasierten Methoden (Sun et al., 2010). Unter diesen verschiedenen Ansätzen hat die pyrosequenzierungsbasierte Methode das umfassendste Mikrobiota-Profil von Koumiss bereitgestellt, unabhängig von phänotypischen Merkmalen und Problemen der Kultivierbarkeit der einzelnen Mikroben. Das Spektrum der funktionellen Gene, die von den Koumiss-Bakterien kodiert werden, und ihre Fermentationskapazitäten sind jedoch nach wie vor schlecht charakterisiert, insbesondere für die seltenen mikrobiellen Populationen.
Die vorliegende Studie verwendete die Einzelzell-Genomik-Technik, um die bakteriellen Metagenome von 10 Koumiss-Proben aus der Mongolei und der Inneren Mongolei Chinas zu analysieren. Die aktuellen Arbeiten haben modernste Technologien zur Untersuchung der Bakterienvielfalt in Milchprodukten eingesetzt. Unsere Arbeit hat die Machbarkeit der Entdeckung von Taxa mit geringer Häufigkeit durch Anwendung des Einzelzell-Metagenomik-Ansatzes gezeigt. Die ermutigenden Ergebnisse würden die Entwicklung und Anwendung neuartiger Ansätze zur Bewältigung von Problemen in einem traditionellen Forschungsgebiet fördern.
Materialien und Methoden
Probenvorbereitung
Für die Metagenomics-Studie wurden insgesamt 10 Koumiss-Proben aus der Mongolei (MG14, MG15, MG16, MG17 und MG18) und der Inneren Mongolei Chinas (NM17, NM18, NM19, NM20 und NM21) entnommen. Die Proben wurden aseptisch gesammelt und in Trockeneis transportiert.
Ein Milliliter jeder Probe wurde nach der in Ward et al. (2013) mit einigen Änderungen. Kurz wurden die Proben in einem Eisbad für 3-5 min aufgetaut. Nachdem die Proben geschmolzen waren, wurden sie einer langsamen Zentrifugation unterzogen, um Verunreinigungen und eukaryotische Zellklumpen zu entfernen. Prokaryotische Zellen wurden dann aus den Milchseren durch Zentrifugation bei 13.000 ×g für 15 min pelletiert. Die Pellets wurden erneut in 2 ml phosphatgepufferter Kochsalzlösung (PBS) mit 1% Triton X-100 suspendiert und 2 h bei 37 ° C inkubiert, um alle verbleibenden eukaryotischen Zellen zu lysieren. Anschließend wurden Bakterien durch Zentrifugation bei 13.000 ×g für 15 min pelletiert und die Pellets in 500 µL PBS resuspendiert. Schließlich wurde der Zentrifugationsschritt noch einmal wiederholt, um die Bakterienzellen zu waschen.
Gradientenverdünnung und Multiple-Displacement-Amplifikation
Zum Nachweis der Niederverdrängungsbakterien wurde die von jeder Koumiss-Probe abgeleitete Bakteriensuspension für die nachfolgende Amplifikationsreaktion seriell verdünnt. Die Zellzahl in jeder Probe wurde unter einem Mikroskop (Nikon, Tokio, Japan) unter Verwendung einer Zellzählungskammer (Qiujing, Shanghai, China) grob geschätzt. Der Verdünnungsschritt wurde fortgesetzt, bis die Zellzahl in jeder Bakteriensuspension etwa 100 erreichte. Die Multiple Displacement Amplifikation der verdünnten Zellen wurde unter Verwendung des REPLI-g Single Cell Kit (Qiagen, Germantown, MD, USA) gemäß den Anweisungen des Herstellers durchgeführt.
Bibliothekskonstruktion und Sequenzierung
Amplifizierte DNA wurde zufällig geschert, und die Fragmente von ungefähr 500 bp wurden ausgewählt. Nach dem Bibliotheksbau wurden PerkinElmer LabChip® GX Touch und StepOnePlusTM Real-Time PCR System für die Bibliotheksqualitätsprüfung eingesetzt. Schließlich wurden 125-bp-Lesevorgänge am gepaarten Ende auf der Illumina HiSeq 2500-Plattform gemäß den Anweisungen des Herstellers sequenziert.
Datenanalyse
Sequenzqualitätsprüfung und Filterung
Vom Sequenzer generierte Raw-Lesevorgänge können künstliche Lesevorgänge von Adapterkontaminationen während der Bibliothekskonstruktion enthalten. Daher wurden drei Schritte durchgeführt, um einen qualitativ hochwertigen sauberen Lese-Datensatz zu erhalten: (1) Beseitigung von Lesevorgängen durch Adapterkontamination; (2) Entfernen von Lesevorgängen mit einer durchschnittlichen Punktzahl unter einem Phred-Wert von Q30, der als niedrigster Grenzwert für eine qualitativ hochwertige Basis angesehen wurde; (3) Entfernen von Lesevorgängen mit einem signifikanten Überschuss von „N“ (≥5% des Lesevorgangs). Die Downstream-Analyse basierte auf den sauberen Daten. Darüber hinaus wurde die statistische Basisqualität, bezogen auf Q30 und den GC-Gehalt, berechnet.
Alignments gegen das Wirtsgenom wurden durchgeführt, um die vom Wirt stammenden Kontaminationssequenzen zu entfernen. Alle vom Wirt stammenden Lesevorgänge wurden vor dem weiteren Vergleich mit Referenzsequenzen des Bakteriengenoms (oder Viren) verworfen. Um genauere Ergebnisse zu erhalten, wurde das MEM–Modell Burrows-Wheeler Aligner (BWA) (Version 0.97a) in den Ausrichtungen verwendet (Li und Durbin, 2009).
Taxonomische Zuordnung und Diversität
Zur taxonomischen Zuordnung auf Gattungs- und Artenebene wurde die Web-Software Metaphlan eingesetzt (Segata et al., 2012). Um die Artenvielfalt innerhalb und zwischen den Proben zu vergleichen, analysierten wir die Alpha- und Beta-Vielfalt durch das R-bezogene Paket.
Assemblierung, Genvorhersage und Annotation lesen
Um umfassendere Informationen zu erhalten, haben wir die gescherten Fragmente zu Genomen (Contigs) zusammengesetzt. Aufgrund des Vorhandenseins mehrerer Arten, das in metagenomischen Proben häufig vorkommt, haben wir jedoch die bioinformatische Genomassemblierungsmethode verbessert, die normalerweise für die Analyse einzelner Arten verwendet wird, indem wir SPAdes (Version 3.6.2), interne Skripte und metagenomische Datenbanken integriert haben (Zerbino und Birney, 2008; Nurk et al., 2013).
Die MetaGeneMark-Software wurde zur Genvorhersage der assemblierten Contigs verwendet (Noguchi et al., 2006). Redundante Gene wurden unter Verwendung von CD-HIT mit der Abdeckung von 90 und 95% Identität entfernt (Li und Godzik, 2006; Fu et al., 2012). Die relativen Häufigkeiten der Gene wurden bestimmt, indem hochwertige Sequenzierungslesewerte mit demselben Verfahren auf den Genkatalog ausgerichtet wurden. Die Downstream-Diskrepanzanalyse basierte auf den relativen Häufigkeiten der Gene. Die Genannotation wurde durchgeführt, indem die hochwertigen Sequenzen gegen mehrere öffentliche Datenbanken (nämlich NCBI non-redundant database, NR; Cluster orthologer Proteingruppen, COGs; Kyoto Encyclopedia of Genes and Genomes, KEGG) unter Verwendung von BLAST (Altschul et al., 1997). Eine Domänensuche wurde mit Interproscan durchgeführt (Mulder und Apweiler, 2008).
Nucleotide Sequence Accession Numbers
Die in dieser Studie berichteten Sequenzdaten wurden in der SRA-Datenbank (Accession No.Modell: SRP083102.
Ergebnisse
Versuchsaufbau und Sequenzierung
Zum Nachweis der wenig abundanten Bakterien in Koumiss wurde die Einzelzellamplifikationstechnik zur Analyse der Metagenome der Proben verwendet. Drei Bakteriensuspensionen mit jeweils etwa 100 Zellen wurden aus einer unabhängigen Koumiss-Probe durch serielle Verdünnung gewonnen. Insgesamt wurden 30 verdünnte Suspensionen analysiert. Jede verdünnte Probe erhielt einen anderen Probencode, d. H. Die Probenidentitätsnummer, gefolgt von 1, 2 oder 3, die die drei separaten Verdünnungen repräsentierten. Basierend auf der Prämisse, dass einige seltene Arten in einer der Verdünnungen vorhanden sein werden, wurde eine multiple Verdrängungsamplifikation der Zellen durchgeführt; und für jede Koumiss-Bakteriensuspension wurden etwa 5 GB Daten generiert.
Aus den 10 Koumiss-Proben (insgesamt 30 Bakteriensuspensionen) wurden insgesamt 1.040.323.864 Rohdaten generiert. Die durchschnittliche Anzahl der Reads für jede 100-Zell-Suspension betrug 34.677.462 (ergänzende Tabelle S1). Nach dem Trimmen und Filtern der unqualifizierten Sequenzen erhielten wir 1.018.381.702 saubere Lesewerte für alle Proben (ergänzende Tabelle S1). Die Werte des Shannon-Index, Simpson-Index, Chao1-Index und die Anzahl der beobachteten Arten (Abbildungen 1-4) zeigten, dass die meisten Koumiss-Proben eine hohe bakterielle Biodiversität aufwiesen. Die Shannon-Wiener-Diversitätskurven zeigten, dass die Sequenztiefe für alle Proben ausreichend war (Abbildung 1).
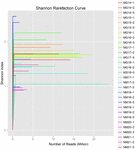
ABBILDUNG 1. Shannon-Verdünnungskurven, die die mikrobielle Vielfalt von Koumiss-Proben abschätzen.
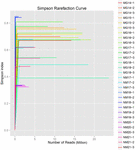
ABBILDUNG 2. Simpson-Verdünnungskurven, die die mikrobielle Vielfalt von Koumiss-Proben abschätzen.
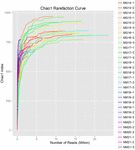
ABBILDUNG 3. Chao1-Verdünnungskurven, die die mikrobielle Diversität von Koumiss-Proben abschätzen.
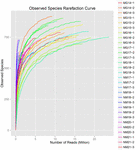
ABBILDUNG 4. Beobachteter Artenverdünnungsindex, der die mikrobielle Vielfalt von Koumiss-Proben abschätzt.
Taxonomische Annotation
Die hochwertigen Sequenzen wurden verschiedenen taxonomischen Ebenen zugeordnet, um eine eingehende Analyse der Bakteriengemeinschaften der Probe zu ermöglichen. Unter Bezugnahme auf einige veröffentlichte Studien zur Biodiversität von Koumiss (Wu et al., 2009; Hao et al., 2010; Sun et al., 2010) klassifizierten wir die bekannten und bisher nicht berichteten Koumiss-assoziierten Bakterien als häufige bzw. seltene Taxa.
Die hochwertigen Sequenzen repräsentierten 13 verschiedene Gattungen (Abbildung 5). Drei von ihnen hatten eine durchschnittliche relative Häufigkeit von über 1%, einschließlich Lactobacillus (L.), Lactococcus und Streptococcus. Insbesondere Lactobacillus und Lactococcus waren die beiden am häufigsten vorkommenden Gattungen im Koumiss. Der Anteil an Lactobacillus in den Proben lag zwischen 52,72 und 99,96%. Zwei Mitglieder dieser Gattung, L. helveticus und L. kefiranofaciens, wurden bei den meisten Koumiss-Proben dominiert (Abbildung 6). Die Art L. buchneri war hauptsächlich in den mongolischen Proben vorhanden, während Lactococcus lactis in den meisten Proben unabhängig von der Probenahmeregion nachgewiesen wurde.
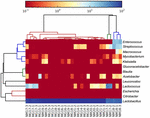
ABBILDUNG 5. Heatmap, die die relative Häufigkeit der in den Koumiss-Proben nachgewiesenen Bakterien auf Gattungsebene zeigt.
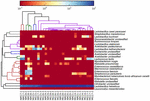
ABBILDUNG 6. Heatmap, die die relative Häufigkeit der in den Koumiss-Proben nachgewiesenen Bakterien auf Artenebene zeigt.
Metagenomische Assemblierung, Genvorhersage und funktionelle Annotation
Die Assemblierung der Reads ergab eine Assemblierungslänge von 614.392.623 bp. Die N50-Werte für die Baugruppen reichten von 5.596 bis 35.200 bp (ergänzende Tabelle S2). Die Anzahl der vorhergesagten Gene in den Koumiss-Proben reichte von 10.347 bis 34.547 mit einer durchschnittlichen Länge von 647,82 bis 985,33 bp (ergänzende Tabelle S3). Obwohl empirische Genfunktionsanalysen den Rahmen der aktuellen Studie sprengten, kommentierten wir die bakteriellen Mikrobiome von Koumiss mithilfe der COG- und KEGG-Datenbanken, die die Genfunktion weitgehend basierend auf der Sequenzhomologie vorhersagten.
Insgesamt 545 Übereinstimmungen in der Annotationsausgabe zeigten eine hohe Homologie zu den Laktoseverwertungsgenen (COGs category of carbohydrate and metabolism, G), und einige von ihnen befanden sich innerhalb derselben Grenze (Tabelle 1).
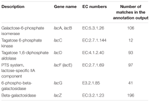
TABELLE 1. Laktosestoffwechsel-verwandte Gene, die im bakteriellen Metagenom von Koumiss annotiert sind.
Einige andere Sequenzen könnten für mutmaßliche Gene innerhalb der COGs-Kategorie des Aminosäuretransports und -metabolismus (E) kodieren, einschließlich Sequenzen, die caseinabbauenden Proteinasen, dem Opp-System zur Aufnahme von Oligopeptiden mit 4-18 Resten und Aminopeptidasen (z. B. Leucylaminopeptidase, Peptidyldipeptidase A, Aminopeptidase N, Prolin-Iminopeptidase, und Endopeptidase). Obwohl einige Sequenzen hohe Ähnlichkeiten mit den für Arginin, Aspartat, Methionin und Isoleucin spezifischen Aminotransferasen aufwiesen, wurde nur eine signifikante Übereinstimmung identifiziert, die einer mutmaßlichen von S. macedonicus stammenden Klasse-I / Klasse-II-Domäne (IPR004839) entsprach -enthaltende Aminotransferase spezifisch für Tyrosin und Phenylalanin. Schließlich teilte eine Anzahl von Sequenzen eine hohe Homologie mit den Aminosäurelyasen, einschließlich S-Ribosylhomocysteinlyase, Argininosuccinatlyase, Aspartat-Ammoniak-Lyase, Cystathionin-Gamma-Lyase, Histidin-Ammoniak-Lyase und O-Acetylhomoserin (Thiol) -Lyase.
Koumiss ist ein beliebtes traditionelles fermentiertes Milchprodukt in der Mongolei und der Inneren Mongolei Chinas. Obwohl eine Reihe von Studien durchgeführt wurden, um die bakterielle Vielfalt in Koumiss zu untersuchen, wurden nur wenige Informationen über die genetische Kapazität der Koumiss-Mikrobiota erhalten. Hier haben wir die Einzelzellamplifikationstechnik angewendet, um die bakteriellen Metagenome von Koumiss zu analysieren, wobei wir uns insbesondere auf die Bakterienpopulation mit geringer Häufigkeit konzentrierten.
Der typische metagenomische Ansatz wurde zuvor angewendet, um die Koumiss-Mikrobiota zu beschreiben. Aufgrund der hohen Kosten für das Erreichen einer tiefen Sequenz wird die seltene Mikrobiota-Population in den Proben jedoch oft unzureichend abgedeckt. Somit bleiben sowohl die phylogenen als auch die funktionellen Metagenome der Minderheitsbakterien begrenzt. Unsere Einzelzellamplifikationsmethode beinhaltete die serielle Verdünnung von Proben zu 100-Zellsuspensionen. Aufgrund der geringen Anzahl von Zellen, die in den verdünnten Koumiss-Proben vorhanden sind, würde nur eine begrenzte Menge an DNA extrahiert. Der Amplifikationsschritt erhöhte die Menge der zu analysierenden DNA-Materialien und erleichterte somit die Metagenomanalyse von Proben, die winzige DNA-Mengen enthielten. Ein Nachteil dieser Methode war die Schwierigkeit, sicherzustellen, dass Sequenzen jedes Taxons dabei gleichermaßen amplifiziert werden; daher konnten die hier erhaltenen Ergebnisse nur die relativen Häufigkeiten von Sequenzen widerspiegeln, nicht jedoch die absoluten Mengen identifizierter Taxa oder funktioneller Gene. Dies hätte jedoch nur geringe Auswirkungen auf unsere Analyse, da sich die vorliegende Studie von anderen veröffentlichten Arbeiten in der Konzentration auf die seltene Bakterienpopulation unterscheidet. Durch diese Arbeit generierte Daten liefern ergänzende Informationen für die unterrepräsentierte Bevölkerung. Wir glauben, dass der vorliegende Ansatz für die zukünftige Analyse der Bakterienvielfalt für verschiedene Arten von ökologischen Umgebungen geeignet ist.
Die bakterielle Mikrobiota von Koumiss besteht hauptsächlich aus Milchsäurebakterien (LABOR) und einigen Essigsäurebakterien (Zhang und Zhang, 2011). Wie erwartet enthielt unser Datensatz hauptsächlich Sequenzen, die die LABOR- und Essigsäurebakterien repräsentierten. Sequenzen, die Lactobacillus helveticus entsprachen, dominierten über alle Proben hinweg. Außerdem Sequenzen, die die Art L. kefiranofaciens darstellen. In: L. buchneri. L. kefiranofaciens. Enterococcus (E.) casseliflavus. E. faecalis. E. faecium. Leuconostoc mesenteroides. Lactococcus lactis und Acetobacter pasteurianus wurden ebenfalls gefunden. Die Identifizierung von Sequenzen verschiedener Taxa reicht möglicherweise nicht aus, um die Lebensfähigkeit der Bakterien zu zeigen, sie spiegelt jedoch die Bakteriengemeinschaft zu einem bestimmten Zeitpunkt des Fermentationsprozesses wider. Miyamoto et al. (2010) legt nahe, dass die bakterielle Prävalenz in den fermentierten Endprodukten mit ihrer Säurestresstoleranz zusammenhängt. Im Allgemeinen haben Laktobazillen eine höhere Säuretoleranz als Laktokokken, was die hohe relative Häufigkeit von Laktobazillen-Sequenzen in unserem Datensatz erklären kann. Andererseits ist das häufige Auftreten von L. helveticus in koumiss fiel mit der Beobachtung der Dominanz von L. helveticus-Sequenzen zusammen.
In unserem Datensatz entsprachen einige Sequenzen L. otakiensis, einer seltenen Art, die in koumiss oder anderen Milchnischen noch nie gemeldet wurde. Die Art wurde zunächst beschrieben und aus der nicht gesalzenen Beizlösung isoliert, die zur Herstellung von Sunki, einer traditionellen japanischen Gurke, verwendet wurde (Watanabe et al., 2009). Es wurde durch Amplified Fragment length polymorphism Profiling basierend auf dem recA-Gen entdeckt. Seitdem wurde nicht berichtet, dass diese Art mit anderen lebensmittelbezogenen Nischen in Verbindung gebracht wird. Daher ist es wahrscheinlich, dass dieses Bakterium zur autochthonen Flora von Gurken gehört. Wir können jedoch nicht ausschließen, dass es einfach aufgrund der Empfindlichkeit der verwendeten Nachweismethode nicht gefunden wurde. Lactobacillus otakiensis kann d-verzweigtkettige Aminosäuren wie d-Leucin, d-allo-Isoleucin und d-Valin produzieren (Doi et al., 2013). Es hat das Potenzial, die Produktionseigenschaften bestimmter fermentierter Lebensmittel zu verbessern (Kato et al., 2011).
Unsere Studie identifizierte auch Sequenzen, die die Art S. macedonicus repräsentieren, die nie als Koumiss-assoziiertes Bakterium gemeldet wurde. Stattdessen ist es eine Starterkultur, die in griechischen Schaf- und Ziegenkäsesorten vorhanden ist (Georgalaki et al., 2000). Einige Mitglieder dieser Spezies sind in der Lage, Exopolysaccharide, Bakteriocine (Vincent et al., 2001; Anastasiou et al., 2015) und Gamma-Aminobuttersäure (Franciosi et al., 2015). Obwohl diese Art häufig aus fermentierten Lebensmitteln isoliert wird, war die ursprüngliche Nische von S. macedonicus umstritten (Guarcello et al., 2016). Erst vor kurzem, Papadimitriou et al. (2015) identifizierten ein erworbenes Plasmid, pSMA198, aus Lactococcus lactis. Das Plasmid wurde wahrscheinlich über ein genetisches Austauschereignis der Vorfahren in einer Milchproduktumgebung übertragen, was auf den Milchursprung von S. macedonicus hindeutet (Papadimitriou et al., 2015). Ähnlich wie S. thermophilus. S. macedonicus ist eng mit den Kommensalen und opportunistischen Erregern des S. bovis / S. equinus-Komplexes verwandt.
Die Identifizierung von Sequenzen, die die Spezies Ruminococcus torques repräsentieren, war unerwartet, da dieses Bakterium normalerweise in der Darmumgebung vorkommt. Es ist eine normale menschliche Darmmikrobe, die Mucinoligosaccharide durch konstitutive Produktion von sekretorischen Glykosidasen abbauen kann (Hoskins et al., 1985). Jüngste klinische Beweise zeigen, dass die fäkale Häufigkeit dieser Spezies bei Kindern mit Autismus-Spektrum-Störung verändert ist; Seine Rolle bei der Störung bleibt jedoch unklar (Wang et al., 2013).
Überraschenderweise stellten einige der Sequenzen potentielle Pathogene dar. Zum Beispiel ist Klebsiella-Pneumonie ein opportunistischer Humanpathogen, der in etwa 40% der menschlichen und tierischen Eingeweide vorkommt. Mycobacterium orygis, früher Oryx-Bazillus genannt, ist ein Mitglied des Mycobacterium tuberculosis-Komplexes, der Tuberkulose beim Menschen verursachen kann (Dawson et al., 2012). Diese beiden Arten wurden in Rohmilch berichtet, aber nicht in Koumiss. Daher könnte ihr Vorhandensein auf die Kontamination während der konventionellen Koumiss-Produktion zurückzuführen sein, insbesondere unter nicht- oder niedrig-aseptischen Manipulationsbedingungen.
Der bakterielle Stoffwechsel spielt eine wichtige Rolle bei der Bildung der Koumiss-Eigenschaften und -Qualität. Mikrobielle Prozesse wie Lipolyse und Proteolyse sind für die Synthese von Koumiss-Aroma- und Geschmacksstoffen erforderlich (Gesudu et al., 2016). Konsistent enthielt das bakterielle Metagenom Sequenzen, die möglicherweise für den Laktoseabbau und proteolytische Systeme kodieren. Im Gegensatz zu den relativ einfachen Laktose-Abbauwegen bestehen die proteolytischen Systeme des Labors aus einer Vielzahl von Enzymen (Chen et al., 2014). Im Vergleich zu anderen nachgewiesenen Koumiss-ARTEN zeichnet sich die dominante Spezies L. helveticus durch eine hohe proteolytische Aktivität aus (Zhang et al., 2015). Die meisten BAKTERIEN besitzen nur eine Zellhüllproteinase, die die Milchkaseinhydrolyse initiiert, während L. helveticus mindestens zwei dieser Enzyme enthält, nämlich PrtH und PrtH2 (Zhao et al., 2011). So können die hohen Anteile an Sequenzen, die den starken proteolytischen Spezies von L. helveticus und Proteolyse-verwandten Genen entsprechen, mit den relativ hohen Gehalten an Peptiden und freien Aminosäuren in Koumiss zusammenhängen.
Eine Schwierigkeit bei der industriellen Koumiss-Produktion ist die Kontrolle der Geschmackswahrnehmung, da Koumiss traditionell durch natürliche Fermentation hergestellt wurde. Daher war es schwierig, den Geschmack und das Profil der wichtigsten Geschmackskomponenten zu definieren, insbesondere in Gegenwart der natürlichen Verunreinigungen. Die Produktion von Schlüsselaromakomponenten ist ein Ergebnis des fermentativen und enzymatischen Abbaus von Aminosäuren, wie den verzweigtkettigen Aminosäuren, Methionin und aromatischen Aminosäuren (Ardo, 2006). Beispiele für Aromakomponenten umfassen Aldehyde, organische Säuren und Ester, die durch den Transaminase (AT)-Weg gebildet werden (Helinck et al., 2004). Dieser Weg wird durch eine Transaminase initiiert, die die Umwandlung einer Aminosäure in ihre entsprechende α-Ketosäure katalysiert (Helinck et al., 2004). Einige Sequenzen in unserem Datensatz entsprachen mutmaßlichen verzweigtkettigen und aromatischen Aminosäure-Aminotransferasen. Insbesondere fanden wir eine mutmaßliche aromatische Aminosäure Aminotransferase I im Contig der Koumiss-assoziierten Spezies, S. macedonicus. Da unsere aktuelle Arbeit nur das Mikrobiom in silico annotierte, reichen unsere Daten nicht aus, um zu zeigen, dass diese identifizierten Gensequenzen tatsächlich funktional waren, um die oben genannten Koumiss-Geschmacksverbindungen herzustellen. Das Vorhandensein dieser Gene deutet jedoch darauf hin, dass sie einige mögliche Kandidaten für solche fermentativen Aktivitäten sind; Dennoch wären weitere experimentelle Arbeiten erforderlich, um ihre genauen funktionellen Rollen aufzuklären.
Darüber hinaus fanden wir Sequenzen, die anderen Aminosäure-Umwandlungswegen entsprechen, einschließlich Aminosäure-Lyasen und Threonin-Aldolase. Ersteres Enzym katalysiert die Umwandlung von Methionin zu Methanthiol (Irmler et al., 2008), was zur Bildung von Dimethyldisulfid und Dimethyltrisulfid führt (Fernandez et al., 2000), während letzteres die Umwandlung von Threonin zu Glycin und Acetaldehyd katalysiert (Ott et al., 2000). Ebenso kann die bloße Lokalisierung dieser Sequenzen innerhalb des Koumiss-Mikrobioms nicht als eindeutiger Beweis für ihre tatsächlichen biologischen Rollen dienen; Zukünftige experimentelle Bestätigung wird erforderlich sein.
Schlussfolgerung
Die vorliegende Studie verwendete eine modifizierte metagenomische Methode zur Analyse des bakteriellen Mikrobioms von Koumiss-Proben, die in der Mongolei und in der Inneren Mongolei Chinas gesammelt wurden. Wir haben sowohl die phylogenen als auch die funktionellen Metagenome der seltenen Arten in Koumiss charakterisiert; und unser Datensatz spiegelt Merkmale wider, die für die Biotechnologie von Interesse und Potenzial sind. Unsere Studie hat zum ersten Mal die Machbarkeit der Einbeziehung der Einzelzell-Amplifikationstechniken beim Nachweis von Koumiss-Bakterien Mikrobiota sowie mikrobielle Kontaminanten. Die hierin entwickelten Techniken können in zukünftigen Studien verwendet werden, um Veränderungen im Koumiss-Mikrobiom entlang des Fermentationsprozesses zu überwachen, wobei der Schwerpunkt auf der mikrobiellen Minderheitenpopulation liegt. Darüber hinaus können andere Omics-Techniken, wie Transkriptomik, Metabolomik, an die aktuelle metagenomische Analyse gekoppelt werden, um die Funktionen und metabolische Kapazität des Koumiss-Mikrobioms zu bestätigen.
Technisch gesehen wäre der nächste Schritt dieser Arbeit die Optimierung der aktuellen Methode. Beispielsweise kann durch Erhöhen der Probenverdünnung vor der DNA-Amplifikation die Chance, seltene und neuartige Bakterien aufzudecken, verbessert werden. Auf der anderen Seite kann eine alternative Sequenzierungstechnik, die lange Lesevorgänge erzeugen kann, wie die Einzelmolekül-Echtzeitsequenzierungstechnologie von Pacific Biosciences, eingesetzt werden, um den Genomassemblierungsprozess zu verbessern.
Autorenbeiträge
WZ und HZ gestalteten die Studie. WZ, GY, JY und L-YK schrieben das Manuskript. QH, WH, WL, BM und TS führten Experimente durch. WZ und QH analysierten Daten. Alle Autoren haben das Manuskript überprüft.
Interessenkonflikterklärung
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Anerkennung
Diese Forschung wurde von der National Natural Science Foundation of China unterstützt (Grant No. 31571815).
Ergänzungsmaterial
Das Ergänzungsmaterial zu diesem Artikel finden Sie online unter: https://www.frontiersin.org/article/10.3389/fmicb.2017.00165/full#supplementary-material
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST und PSI-BLAST: eine neue Generation von Proteindatenbank-Suchprogrammen. Nukleinsäuren Res. 25, 3389-3402. doi: 10.1093/nar/25.17.3389
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Anastasiou, R., Driessche, G. V., Boutou, E., Kazou, M., Alexandraki, V., Vorgias, C. E., et al. (2015). Konstruierte Stämme von Streptococcus macedonicus in Richtung eines osmotischen stressresistenten Phänotyps behalten ihre Fähigkeit, das Bakteriocin macedocin unter hyperosmotischen Bedingungen zu produzieren. In: J. Biotechnol. 212, 125–133. doi: 10.1016/j.jbiotec.2015.08.018
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Ardo, Y. (2006). Geschmacksbildung durch Aminosäurekatabolismus. In: Biotechnol. Adv. 24, 238-242. doi: 10.1016/j.biotech.2005.11.005
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitation Chen, Y. F., Zhao, W. J., Wu, R. N., Sun, Z. H., Zhang, W. Y., Wang, J. C., et al. (2014). Proteomanalyse von Lactobacillus helveticus H9 während des Wachstums in Magermilch. J. Molkerei Sci. 97, 7413–7425. Ursprungsbezeichnung: 10.3168/jds.2014-8520
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Dawson, KL, Bell, A., Kawakami, RP, Coley, K., Yates, G. und Collins, DM (2012). Übertragung von Mycobacterium orygis (M. tuberculosis complex species) von einem Tuberkulose-Patienten auf eine Milchkuh in Neuseeland. J. Clin. Microbiol. 50, 3136–3138. doi: 10.1128/JCM.01652-12
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Doi, K., Mori, K., Mutaguchi, Y., Tashiro, K., Fujino, Y., Ohmori, T., et al. (2013). Entwurf einer Genomsequenz des D-verzweigtkettigen Aminosäureproduzenten Lactobacillus otakiensis JCM 15040T, isoliert aus einer traditionellen japanischen Gurke. Genom Announc. 1, e546-e513. doi: 10.1128/genomeA.00546-13
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Fernandez, M., van Doesburg, W., Rutten, G. A., Marugg, J. D., Alting, A. C., van Kranenburg, R., et al. (2000). Molekulare und funktionelle Analysen des metC-Gens von Lactococcus lactis, das für Cystathionin-Beta-Lyase kodiert. Appl. ENVIRON. Microbiol. 66, 42–48. modell: 10.1128/AEM.66.1.42-48.2000
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Franciosi, E., Carafa, I., Nardin, T., Schiavon, S., Poznanski, E., Cavazza, A., et al. (2015). Biodiversität und Gamma-Aminobuttersäureproduktion durch Milchsäurebakterien, die aus traditionellen alpinen Kuhmilchkäse isoliert wurden. Biomed Res. Int. 2015, 625740. doi: 10.1155/2015/625740
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Fu, L., Niu, B., Zhu, Z., Wu, S. und Li, W. (2012). CD-HIT: beschleunigt für das Clustern der Sequenzierungsdaten der nächsten Generation. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
PubMed Abstract | CrossRef Full Text | Google Scholar
Georgalaki, M. D., Sarantinopoulos, P., Ferreira, E. S., De Vuyst, L., Kalantzopoulos, G., and Tsakalidou, E. (2000). Biochemical properties of Streptococcus macedonicus strains isolated from Greek Kasseri cheese. J. Appl. Microbiol. 88, 817–825. doi: 10.1046/j.1365-2672.2000.01055.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Gesudu, Q. M., Zheng, Y., Xi, X. X., Hou, Q. C., Xie, H. Y., Huang, W. Q., et al. (2016). Untersuchung der Bakterienpopulationsstruktur und -dynamik in traditionellen Koumiss aus der Inneren Mongolei mittels Einzelmolekül-Echtzeitsequenzierung. J. Molkerei Sci. 99, 7852–7863. Ursprungsbezeichnung: 10.3168/jds.2016-11167
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitation Guarcello, R., Gaglio, R., Caggia, C., Rapisarda, T., et al. (2016). Eine großflächige Anwendung ausgewählter autochthoner Milchsäurebakterien für die Herstellung von Pecorino Siciliano-Käse mit g.U. Lebensmittel Microbiol. 59, 66–75. geburtsdatum: 10.1016/j.fm.2016.05.011
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Zhao, Y., Zhao, L., Zhang, H., Zhai, Z., Huang, Y., Liu, X., et al. (2010). Identifizierung der bakteriellen Biodiversität in Koumiss durch denaturierende Gradientengelelektrophorese und spezies-spezifische Polymerase-Kettenreaktion. J. Molkerei Sci. 93, 1926–1933. Ursprungsbezeichnung: 10.3168/jds.2009-2822
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Helinck, S., Le Bars, D., Moreau, D. und Yvon, M. (2004). Fähigkeit thermophiler Milchsäurebakterien, Aromastoffe aus Aminosäuren herzustellen. Appl. ENVIRON. Microbiol. 70, 3855–3861. modell: 10.1128/AEM.70.7.3855-3861.2004
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Hoskins, LC, Agustines, M., McKee, WB, Boulding, ET, Kriaris, M. und Niedermeyer, G. (1985). Mucinabbau in menschlichen Kolonökosystemen. Isolierung und Eigenschaften von fäkalen Stämmen, die ABH-Blutgruppenantigene und Oligosaccharide aus Mucin-Glykoproteinen abbauen. J. Clin. Investieren. 75, 944–953. doi: 10.1172/JCI111795
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Irmler, S., Raboud, S., Beisert, B., Rauhut, D. und Berthoud, H. (2008). Klonierung und Charakterisierung von zwei Lactobacillus casei-Genen, die für eine Cystathionin-Lyase kodieren. Appl. ENVIRON. Microbiol. 74, 99–106. modell: 10.1128/AEM.00745-07
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Jagielski, V. (1877). Der Wert von Koumiss bei der Behandlung von Übelkeit, Erbrechen und Unfähigkeit, andere Nahrung auf dem Magen zu behalten. Br. Med. J. 2, 919-921. doi: 10.1136/bmj.2.887.919
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Kato, S., Ishihara, T., Hemmi, H., Kobayashi, H. und Yoshimura, T. (2011). Veränderungen der D-Aminosäurekonzentrationen und der mikrobiellen Gemeinschaftsstrukturen während der Gärung von Rot- und Weißweinen. In: J. Biosci. In: Bioeng. 111, 104–108. doi: 10.1016/j.jbiosc.2010.08.019
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Li, H. und Durbin, R. (2009). Schnelle und genaue kurze Leseausrichtung mit Burrows-Wheeler Transform. Bioinformatik 25, 1754-1760. doi: 10.1093/bioinformatik/btp324
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Li, W. und Godzik, A. (2006). Cd-hit: ein schnelles Programm zum Clustern und Vergleichen großer Mengen von Protein- oder Nukleotidsequenzen. Bioinformatik 22, 1658-1659. doi: 10.1093/bioinformatik/btl158
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitation Miyamoto, M., Seto, T., Nakajima, H., Gombojav, A., et al. (2010). Denaturierende Gradientengelelektrophoreseanalyse von Milchsäurebakterien und Hefen in traditioneller mongolischer fermentierter Milch. Lebensmittel Sci. Technol. 16, 319-326. doi: 10.3136/fstr.16.319
CrossRef Volltext / Google Scholar
Mulder, NJ und Apweiler, R. (2008). Die InterPro Datenbank und Werkzeuge für die Proteindomänenanalyse. Curr. Ja. Bioinformatik Kapitel 2, Unit2.7. doi: 10.1002/0471250953.bi0207s21
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Noguchi, H., Park, J. und Takagi, T. (2006). MetaGene: prokaryotische Genfindung aus Umweltgenom-Shotgun-Sequenzen. Nukleinsäuren Res. 34, 5623-5630. doi: 10.1093/nar/gkl723
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A. A., Korobeynikov, A., Lapidus, A., et al. (2013). Assemblierung von einzelligen Genomen und Mini-Metagenomen aus chimären MDA-Produkten. In: J. Comput. Biol. 20, 714–737. doi: 10,1089/cmb.2013.0084
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Ott, A., Germond, J. E., und Chaintreau, A. (2000). Ursprung von Acetaldehyd während der Milchfermentation unter Verwendung von (13) C-markierten Vorläufern. J. Agric. Lebensmittel Chem. 48, 1512–1517. doi: 10.1021/jf9904867
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Pan, DD, Zeng, XQ und Yan, YT (2011). Charakterisierung von Lactobacillus fermentum SM-7 isoliert aus Koumiss, einem potenziellen probiotischen Bakterium mit cholesterinsenkender Wirkung. J. Sci. Lebensmittel. Agric. 91, 512–518. doi: 10.1002/jsfa.4214
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Papadimitriou, K., Anastasiou, R., Maistrou, E., Plakas, T., Papandreou, N. C., Hamodrakas, S. J., et al. (2015). Der Erwerb durch horizontalen Gentransfer des Plasmids pSMA198 durch Streptococcus macedonicus ACA-DC 198 weist auf den Milchursprung der Art hin. Plus EINS 10:e0116337. doi: 10.1371/Zeitschrift.pone.0116337
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Segata, N., Waldron, L., Ballarini, A., Narasimhan, V., Jousson, O. und Huttenhower, C. (2012). Metagenomische mikrobielle Gemeinschaftsprofilierung unter Verwendung einzigartiger kladenspezifischer Markergene. Nat. Methoden 9, 811-814. doi: 10.1038/nmeth.2066
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Zhang, J., Liu, W.,Zhang, J., Yu, J., Zhang, W., Cai, C., et al. (2010). Identifizierung und Charakterisierung der aus Koumiss in China isolierten dominanten Laktobazillen. In: J. Gen. Appl. Microbiol. 56, 257–265. artikelnummer: 10.2323/jgam.56.257
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Thompson, J. (1879). Der Wert von Koumiss bei der Verschwendung von Krankheiten. Br. Med. J. 1, 270. doi: 10.1136/bmj.1.1466.270-c
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Vincent, SJ, Faber, EJ, Neeser, JR, Stingele, F. und Kamerling, JP (2001). Struktur und Eigenschaften des von Streptococcus macedonicus Sc136 produzierten Exopolysaccharids. Glykobiologie 11, 131-139. doi: 10.1093/glycob/11.2.131
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Wang, L., Christophersen, C. T., Sorich, M. J., Gerber, J. P., Angley, M. T. und Conlon, M. A. (2013). Erhöhte Häufigkeit von Sutterella spp. und Wiederkäuerdrehmomente im Kot von Kindern mit Autismus-Spektrum-Störung. Mol. Autismus 4, 42. doi: 10.1186/2040-2392-4-42
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Ward, TL, Hosid, S., Ioshikhes, I. und Altosaar, I. (2013). Human milk metagenome: eine Analyse der funktionellen Kapazität. BMC Microbiol. 13:116. doi: 10.1186/1471-2180-13-116
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Watanabe, K., Fujimoto, J., Tomii, Y., Sasamoto, M., Makino, H., Kudo, Y., et al. (2009). Lactobacillus kisonensis sp. Nov., Lactobacillus otakiensis sp. Nov., Lactobacillus rapi sp. Nov. und Lactobacillus sunkii sp. Nov., heterofermentative Spezies, isoliert aus Sunki, einer traditionellen japanischen Gurke. Int. In: J. Syst. In: Evol. Microbiol. 59(Pt 4), 754-760. Ursprungsbezeichnung: 10.1099/ijs.0.004689-0
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Wu, R., Wang, L., Wang, J., Li, H., Menghe, B., Wu, J., et al. (2009). Isolierung und vorläufige probiotische Selektion von Laktobazillen aus Koumiss in der Inneren Mongolei. In : J. Basic Microbiol. 49, 318–326. doi: 10.1002/jobm.200800047
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Zerbino, Dr. und Birney, E. (2008). Velvet: Algorithmen für de novo Short Read Assembly mit de Bruijn Graphen. Genome Res. 18, 821-829. Ursprungsbezeichnung: 10.1101/gr.074492.107
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Zhang, W., Sun, Z., Sun, T. und Zhang, H. (2010a). PCR-Screening und Sequenzanalyse von iol-Clustern in aus Koumiss isolierten Lactobacillus casei-Stämmen. Folia Microbiol. (Prag) 55, 603-606. doi: 10.1007/s12223-010-0097-3
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Zhang, W., Yu, D., Sun, Z., Wu, R., Chen, X., Chen, W., et al. (2010b). Vollständige Genomsequenz von Lactobacillus casei Zhang, einem neuen probiotischen Stamm, der aus traditionellem hausgemachtem Koumiss in der Inneren Mongolei, China, isoliert wurde. In: J. Bacteriol. 192, 5268–5269. Ursprungsbezeichnung: 10.1128/JB.00802-10
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Zhang, W. und Zhang, H. (2011). „Fermentation und Koumiss“, in Handbuch der Fermentationstechnologie für Lebensmittel und Getränke, 2nd Edn, ed. Y. H. Hui (Boca Raton, FL: CRC Press), 165-172.
Google Scholar
Zhang, WY, Chen, YF, Zhao, WJ, Kwok, LY und Zhang, HP (2015). Genexpression des proteolytischen Systems von Lactobacillus helveticus H9 während der Milchfermentation. Ann. Microbiol. 65, 1171–1175. Ursprungsbezeichnung: 10.3168/jds.2014-8520
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Zhao, W., Chen, Y., Sonne, Z., Wang, J., Zhou, Z., Sonne, T., et al. (2011). Vollständige Genomsequenz von Lactobacillus helveticus H10. In: J. Bacteriol. 193, 2666–2667. Ursprungsbezeichnung: 10.1128/JB.00166-11
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar