- Introdução
- Materiais e Métodos
- Preparação de Amostras
- diluição de gradiente e amplificação de deslocamento múltiplo
- Construção e sequenciamento da Biblioteca
- Análise de dados
- verificação e filtragem da qualidade da sequência
- atribuição e diversidade taxonômica
- leia montagem, previsão de genes e anotação
- números de adesão de sequência de nucleotídeos
- resultados
- delineamento Experimental e sequenciamento
- anotação taxonômica
- montagem metagenômica, predição gênica e anotação funcional
- discussão
- conclusão
- contribuições do autor
- Declaração de conflito de interesses
- reconhecimento
- Material Suplementar
Introdução
Koumiss, também chamado de chige, chigo, arrag, ou airag, na língua mongol, é um tipo tradicional de produtos lácteos fermentados. Tem sido um alimento popular na Mongólia E Na mongólia interior da China há séculos (Zhang e Zhang, 2011). As pessoas nessas regiões costumavam consumir koumiss durante grandes festividades e ofertas de sacrifício (Zhang e Zhang, 2011). O registro mais antigo da produção de koumiss pode ser rastreado até a Dinastia Han (BC202-AD202). Este produto alcançou popularidade generalizada durante a Dinastia Yuan (AD1271-AD1368) (Zhang et al., 2010b). Hoje em dia, koumiss é um alimento comum para a população local da Mongólia E Da mongólia interior, embora apenas em poucas dessas áreas seja fabricado em escala industrial. Koumiss não apenas fornece nutrientes ricos, incluindo alto conteúdo de aminoácidos essenciais e vitaminas, mas também acredita-se que alivia uma ampla gama de condições médicas e é benéfico para o cuidado pós-operatório (Jagielski, 1877; Thompson, 1879).Tradicionalmente, koumiss é comumente produzido em barris de madeira, recipientes feitos de pele de animal ou urnas. A fermentação ocorre naturalmente à temperatura ambiente após a adição de leite de égua filtrado no recipiente com koumiss antigo, que serve como cultura inicial (Zhang e Zhang, 2011). Koumiss é uma boa fonte de novas bactérias do potencial biotecnológico (Zhang et al., 2010a; Pan et al., 2011). Portanto, é de intenso interesse explorar e preservar o máximo possível de bactérias koumiss associadas à fermentação. Durante as últimas décadas, vários estudos foram realizados para investigar a comunidade bacteriana de koumiss (Wu et al., 2009; Hao et al., 2010), estudado principalmente por métodos baseados em cultura, Biologia molecular e pirosequenciamento (Sun et al., 2010). Entre essas diferentes abordagens, o método baseado em pirosequenciamento forneceu o perfil de microbiota mais abrangente de koumiss independente de características fenotípicas e problemas de cultivabilidade dos micróbios individuais. No entanto, o espectro de genes funcionais codificados pela bactéria koumiss e suas capacidades fermentativas permanecem mal caracterizados, particularmente para as raras populações microbianas.
o presente estudo utilizou a técnica de genômica unicelular para analisar os metagenomas bacterianos de 10 amostras de koumiss coletadas da Mongólia e mongólia interior da China. O trabalho atual aplicou tecnologias de ponta na investigação da diversidade bacteriana em produtos lácteos. Nosso trabalho demonstrou a viabilidade de descobrir táxons de baixa abundância aplicando a abordagem de metagenômica de célula única. Os resultados encorajadores promoveriam o desenvolvimento e a aplicação de novas abordagens no combate aos problemas num campo de investigação tradicional.
Materiais e Métodos
Preparação de Amostras
Um total de 10 koumiss foram coletadas amostras de Mongólia (MG14, MG15, MG16, MG17, e MG18) e Mongólia Interior da China (NM17, NM18, NM19, NM20, e NM21) para o metagenomics estudo. As amostras foram coletadas assepticamente e transportadas em gelo seco.
um mililitro de cada amostra foi pré-tratado de acordo com a metodologia descrita em Ward et al. (2013) com algumas modificações. Resumidamente, as amostras foram descongeladas em banho-maria por 3-5 min. Depois que as amostras derreteram, elas foram submetidas a centrifugação de baixa velocidade para remover impurezas e aglomerados de células eucarióticas. As células procarióticas foram então granuladas dos soros de leite por centrifugação a 13.000 × g por 15 min. Os pellets foram re-suspensos em 2 mL de solução salina tamponada com fosfato (PBS) com Triton X-100 a 1% e incubados por 2 h a 37°C para lise quaisquer células eucarióticas restantes. Posteriormente, as bactérias foram peletizadas por centrifugação a 13.000 × g por 15 min e os pellets foram re-suspensos em 500 µL PBS. Finalmente, a etapa de centrifugação foi repetida mais uma vez para lavar as células bacterianas.
diluição de gradiente e amplificação de deslocamento múltiplo
para detectar as bactérias de baixa abundância, a suspensão bacteriana derivada de cada amostra de koumiss foi diluída em série para posterior reação de amplificação. O número de células em cada amostra foi aproximadamente estimado sob um microscópio (Nikon, Tóquio, Japão) usando uma câmara de contagem de células (Qiujing, Xangai, China). A etapa de diluição foi continuada até que o número de células em cada suspensão bacteriana atingisse aproximadamente 100. A amplificação de deslocamento múltiplo das células diluídas foi realizada usando o kit de célula única REPLI-g (Qiagen, Germantown, MD, EUA) de acordo com as instruções do fabricante.
Construção e sequenciamento da Biblioteca
o DNA amplificado foi cortado aleatoriamente e os fragmentos de aproximadamente 500 PB foram selecionados. Após a construção da biblioteca, o PerkinElmer LabChip ® GX Touch e o StepOnePlusTM real-Time PCR System foram utilizados para inspeção de qualidade da biblioteca. Finalmente, as leituras emparelhadas de 125 bp foram sequenciadas na plataforma Illumina HiSeq 2500 de acordo com as instruções do fabricante.
Análise de dados
verificação e filtragem da qualidade da sequência
as leituras brutas geradas pelo sequenciador podem conter leituras artificiais de contaminação do adaptador durante a construção da biblioteca. Portanto, três etapas foram realizadas para obter um conjunto de dados de leitura limpa de alta qualidade: (1) eliminação de leituras causadas por contaminação do adaptador; (2) remoção de leituras com uma pontuação média abaixo de uma pontuação phred de Q30, que foi considerada como o ponto de corte mais baixo para uma base de alta qualidade; (3) Remoção de leituras com um excesso significativo de “N” (≥5% da leitura). A análise a jusante foi baseada nos dados limpos. Além disso, foram calculadas a qualidade da base estatística, com base no Q30 e no conteúdo do GC.
alinhamentos contra o genoma do hospedeiro foram realizados para remover as sequências de contaminantes originadas pelo hospedeiro. Quaisquer leituras originadas pelo hospedeiro foram descartadas antes de uma comparação adicional com sequências de referência do genoma de bactérias (ou vírus). Para obter resultados mais precisos, foi utilizado o modelo Mem Burrows–Wheeler aligner (BWA) (versão 0.97 a) nos alinhamentos (Li e Durbin, 2009).
atribuição e diversidade taxonômica
o software da Web Metaphlan foi usado para atribuição taxonômica aos níveis de gênero e espécie (Segata et al., 2012). Para comparar a diversidade de espécies dentro e entre as amostras, analisamos a diversidade alfa e beta pelo pacote relacionado ao R.
leia montagem, previsão de genes e anotação
para obter informações mais abrangentes, reunimos os fragmentos cortados em genoma (contigs). No entanto, devido à presença de múltiplas espécies, o que é comum em amostras metagenômicas, melhoramos o método de montagem do genoma da Bioinformática normalmente usado para análise de espécies únicas integrando Espadas (versão 3.6.2), scripts internos e bancos de dados metagenômicos (Zerbino e Birney, 2008; Nurk et al., 2013).
o software MetaGeneMark foi usado para predição gênica dos contigs montados (Noguchi et al., 2006). Genes redundantes foram removidos usando CD-HIT com a cobertura de 90 e 95% de identidade (Li e Godzik, 2006; Fu et al., 2012). As abundâncias relativas dos genes foram determinadas alinhando as leituras de sequenciamento de alta qualidade ao catálogo de genes usando o mesmo procedimento. A análise discrepante a jusante foi baseada nas abundâncias relativas do gene. A anotação gênica foi realizada alinhando as sequências de alta qualidade em vários bancos de dados públicos (ou seja, banco de dados não redundante NCBI, nr; Clusters de grupos Ortólogos de proteínas, COGs; Kyoto Encyclopedia of Genes and Genomes, KEGG) using BLAST (Altschul et al., 1997). Uma pesquisa de domínio foi realizada usando Interproscan (Mulder e Apweiler, 2008).
números de adesão de sequência de nucleotídeos
os dados de sequência relatados neste estudo foram depositados no banco de dados SRA (número de Adesão.SRP083102).
resultados
delineamento Experimental e sequenciamento
para detectar as bactérias de baixa abundância em koumiss, a técnica de amplificação unicelular foi utilizada para analisar os metagenomas das amostras. Três suspensões bacterianas, cada uma de aproximadamente 100 células, foram derivadas de uma amostra independente de koumiss por diluição em série. Foram analisadas 30 suspensões diluídas. Cada amostra diluída recebeu um código de amostra diferente, ou seja, o número de identidade da amostra seguido por 1, 2 ou 3, representando as três diluições separadas. Com base na premissa de que algumas espécies raras estarão presentes em uma das diluições, foi realizada amplificação de deslocamento múltiplo das células; e cerca de 5 Gb de dados foram gerados para cada suspensão bacteriana de koumiss.
um total de 1.040.323.864 leituras brutas foram geradas a partir das 10 amostras de koumiss (um total de 30 suspensões bacterianas). O número médio de leituras para cada suspensão de 100 células foi de 34.677.462 (tabela suplementar S1). Após o corte e filtragem das sequências não qualificadas, obtivemos 1.018.381.702 leituras limpas para todas as amostras (tabela suplementar S1). Os valores de Shannon index, Simpson index, Chao1 index e o número de espécies observadas (figuras 1-4) mostraram que a maioria das amostras de koumiss tinha uma alta biodiversidade bacteriana. As curvas de diversidade de Shannon-Wiener mostraram que a profundidade da sequência foi adequada para todas as amostras (Figura 1).
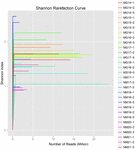
FIGURA 1. Curvas de rarefação de Shannon que estimam a diversidade microbiana de amostras de koumiss.
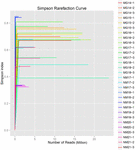
FIGURA 2. Curvas de rarefação de Simpson que estimam a diversidade microbiana de amostras de koumiss.
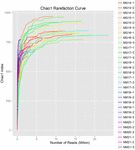
FIGURA 3. Curvas de rarefação Chao1 que estimam a diversidade microbiana de amostras de koumiss.
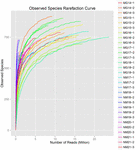
FIGURA 4. Índice de rarefação de espécies observadas que estimam a diversidade microbiana de amostras de koumiss.
anotação taxonômica
as sequências de alta qualidade foram atribuídas a diferentes níveis taxonômicos para permitir uma análise aprofundada das comunidades bacterianas da amostra. Com referência a alguns estudos publicados sobre a biodiversidade de koumiss (Wu et al., 2009; Hao et al., 2010; Sun et al., 2010), classificamos as bactérias conhecidas e anteriormente não relatadas associadas a koumiss como táxons comuns e raros, respectivamente.
as sequências de alta qualidade representaram 13 gêneros diferentes (Figura 5). Três deles tinham uma abundância relativa média de mais de 1%, incluindo Lactobacillus (L.), Lactococcus e Streptococcus. Particularmente, Lactobacillus e Lactococcus foram os dois gêneros mais abundantes encontrados no koumiss. A proporção de Lactobacillus nas amostras variou de 52,72 a 99,96%. Dois membros deste gênero, l. helveticus e L. kefiranofaciens, foram dominados entre a maioria das amostras de koumiss (Figura 6). A espécie L. buchneri estava presente principalmente nas amostras da Mongólia, enquanto Lactococcus lactis foi detectado na maioria das amostras, independentemente da região de amostragem.
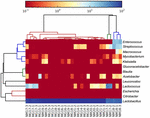
FIGURA 5. Mapa de calor mostrando a abundância relativa de bactérias detectadas nas amostras de koumiss no nível do gênero.
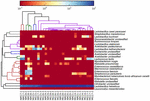
FIGURA 6. Mapa de calor mostrando a abundância relativa de bactérias detectadas nas amostras de koumiss no nível das espécies.
montagem metagenômica, predição gênica e anotação funcional
a montagem das leituras resultou em um comprimento de montagem de 614.392.623 PB. Os valores de N50 para os conjuntos variaram de 5.596 a 35.200 PB (tabela suplementar S2). O número de genes previstos nas amostras de koumiss variou de 10.347 a 34.547, com um comprimento médio variando de 647,82 a 985,33 Pa (tabela suplementar S3). Embora as análises funcionais de genes empíricos estivessem além do escopo do estudo atual, anotamos os microbiomas bacterianos de koumiss usando os bancos de dados COG e KEGG, que previram a função gênica em grande parte com base na homologia de sequência.
um total de 545 correspondências na produção de anotação mostrou alta homologia aos genes de Utilização da lactose (categoria COGs de carboidratos e metabolismo, G), e alguns deles estavam localizados dentro do mesmo contig (Tabela 1).
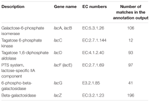
QUADRO 1. Genes relacionados ao metabolismo da Lactose anotados no metagenoma bacteriano de koumiss.
Algumas outras sequências podem código para putativo genes dentro do Cmv categoria de aminoácidos de transporte e metabolismo (E), incluindo as seqüências que correspondeu a caseína degradantes de proteinases, a Opp sistema de assumir os oligopeptídeos de 4-18 resíduos, e aminopeptidases (por exemplo, leucyl aminopeptidase, peptidyl-dipeptidase Um, aminopeptidase N, prolina iminopeptidase, e a endopeptidase). Embora algumas sequências compartilhada de alta semelhanças com as aminotransferases específicos para arginina, aspartato, metionina, e isoleucina, apenas uma significativa correspondência foi identificado, o que correspondeu a um suposto S. macedonicus de origem de classe I/classe II de domínio (IPR004839) contendo aminotransferase específicos de tirosina e fenilalanina. Finalmente, um número de sequências compartilhada alta homologia de aminoácidos lyases, incluindo o S-ribosylhomocysteine liase, argininosuccinate liase, aspartato de amônia-liase, synthase gama-liase, histidina amônia-liase, e O-acetylhomoserine (thiol)-liase.
discussão
Koumiss é um popular produto lácteo fermentado tradicional Na mongólia e Na mongólia interior da China. Embora vários estudos tenham sido realizados para investigar a diversidade bacteriana em koumiss, pouca informação foi obtida sobre a capacidade genética da microbiota de koumiss. Aqui, aplicamos a técnica de amplificação unicelular para analisar os metagenomas bacterianos de koumiss, particularmente com foco na população bacteriana de baixa abundância.
a abordagem metagenômica típica foi aplicada anteriormente para descrever a microbiota de koumiss. No entanto, devido ao alto custo de alcançar uma sequência profunda, a população rara de microbiota nas amostras é frequentemente coberta inadequadamente. Assim, tanto os metagenomas filogênicos quanto funcionais das bactérias minoritárias permanecem limitados. Nosso método de amplificação de célula única envolveu a diluição serial de amostras para suspensões de 100 células. Devido ao baixo número de células presentes nas amostras de koumiss diluídas, apenas uma quantidade limitada de DNA seria extraída. A etapa de amplificação aumentou a quantidade de materiais de DNA a serem analisados e, portanto, facilitou a análise do metagenoma de amostras contendo pequenas quantidades de DNA. Uma desvantagem desse método foi a dificuldade em garantir que as sequências de cada táxon fossem igualmente amplificadas no processo; portanto, os resultados obtidos aqui só poderiam refletir a abundância relativa de sequências, mas não quantidades absolutas de táxons identificados ou genes funcionais. No entanto, isso teria pouco efeito em nossa análise, pois o presente estudo difere de outros trabalhos publicados no foco da população bacteriana rara. Os dados gerados por este trabalho fornecem informações complementares à população sub-representada. Acreditamos que a abordagem atual é adequada para análises futuras da diversidade de bactérias para diferentes tipos de ambientes ecológicos.
a microbiota bacteriana de koumiss consiste principalmente em bactérias do ácido láctico (LAB) e algumas bactérias do ácido acético (Zhang e Zhang, 2011). Como esperado, nosso conjunto de dados continha principalmente sequências representando as bactérias do Laboratório e do ácido acético. Sequências correspondentes a Lactobacillus helveticus estavam dominando todas as amostras. Além disso, sequências representando a espécie, l. kefiranofaciens. L. buchneri. L. kefiranofaciens. Enterococcus (E.) casseliflavus. E. faecalis. E. faecium. Leuconostoc mesenteroides. Lactococcus lactis e Acetobacter pasteurianus também foram encontrados. A identificação de sequências de diferentes táxons pode não ser suficiente para mostrar a viabilidade da bactéria, mas reflete a comunidade bacteriana em algum momento do processo de fermentação. Miyamoto et al. (2010) sugere que a prevalência bacteriana nos produtos fermentados finais está relacionada à sua tolerância ao estresse ácido. Geralmente, os lactobacilos têm maior tolerância ao ácido do que os lactococos, o que pode explicar a alta abundância relativa de sequências de lactobacilos presentes em nosso conjunto de dados. Por outro lado, a ocorrência frequente de L. helveticus em koumiss coincidiu com a observação do domínio das sequências de L. helveticus.
em nosso conjunto de dados, algumas sequências corresponderam a L. otakiensis, que é uma espécie rara que nunca foi relatada em koumiss ou outros nichos lácteos. A espécie foi primeiramente descrita e isolada da solução de decapagem não salgada usada na produção de sunki, um picles Tradicional Japonês (Watanabe et al., 2009). Foi descoberto por polimorfismo de comprimento de fragmento amplificado com base no gene recA. Desde então, esta espécie não foi relatada como associada a outros nichos relacionados a alimentos. Assim, é provável que esta bactéria pertença à flora autóctone dos picles. No entanto, não podemos excluir a possibilidade de não ter sido encontrada simplesmente devido à sensibilidade do método de detecção empregado. Lactobacillus otakiensis pode produzir aminoácidos de cadeia ramificada d, como D-leucina, d-allo-isoleucina e d-valina (Doi et al., 2013). Tem potencial para ser usado na melhoria das características de produção de certos alimentos fermentados (Kato et al., 2011).Nosso estudo também identificou sequências que representam a espécie S. macedonicus, que nunca foi relatada como uma bactéria associada a koumiss. Em vez disso, é uma cultura inicial presente nos queijos gregos de ovelha e cabra (Georgalaki et al., 2000). Alguns membros desta espécie são capazes de produzir exopolissacarídeos, bacteriocinas (Vincent et al., 2001; Anastasiou et al., 2015) e ácido gama-aminobutírico (Franciosi et al., 2015). Embora esta espécie seja frequentemente isolada de alimentos fermentados, o nicho original de S. macedonicus tem sido controverso (Guarcello et al., 2016). Não até recentemente, Papadimitriou et al. (2015) identificaram um plasmídeo adquirido, pSMA198, de Lactococcus lactis. O plasmídeo provavelmente foi transferido por meio de um evento ancestral de troca genética dentro de um ambiente de produtos lácteos, sugerindo a origem láctea de S. macedonicus (Papadimitriou et al., 2015). Semelhante a S. thermophilus. S. macedonicus está intimamente relacionado com os patógenos comensais e oportunistas do complexo S. bovis/S. equinus.
a identificação de sequências que representam a espécie Ruminococcus torques foi inesperada, pois essa bactéria é geralmente encontrada no ambiente intestinal. É um micróbio intestinal humano normal que pode degradar oligossacarídeos de mucina pela produção constitutiva de glicosidases secretoras (Hoskins et al., 1985). Evidências clínicas recentes mostram que a abundância fecal desta espécie é alterada em crianças com transtorno do espectro do autismo; no entanto, seu papel no transtorno permanece incerto (Wang et al., 2013).Surpreendentemente, algumas das sequências representavam potenciais patógenos. Por exemplo, a pneumonia por Klebsiella é um patógeno humano oportunista que reside em cerca de 40% das tripas humanas e animais. Mycobacterium orygis, anteriormente chamado de bacilo oryx, é um membro do complexo Mycobacterium tuberculosis que pode causar tuberculose humana (Dawson et al., 2012). Essas duas espécies foram relatadas em leite cru, mas não koumiss. Portanto, sua presença pode atribuir à contaminação durante a produção convencional de koumiss, especialmente sob condições de manipulação NÃO ou baixa asséptica.
o metabolismo bacteriano desempenha um papel importante na formação das características e qualidade de koumiss. Processos de base microbiana, como lipólise e proteólise, são necessários para sintetizar compostos aromáticos e aromatizantes de koumiss (Gesudu et al., 2016). Consistentemente, o metagenoma bacteriano continha sequências que potencialmente codificam para degradação da lactose e sistemas proteolíticos. Ao contrário das vias catabólicas de lactose relativamente simples, os sistemas proteolíticos de laboratório são compostos por uma gama diversificada de enzimas (Chen et al., 2014). Comparado com outro laboratório koumiss detectado, a espécie dominante l. helveticus é caracterizada por uma alta atividade proteolítica (Zhang et al., 2015). A maioria dos laboratórios possui apenas uma proteinase de envelope celular que inicia a hidrólise da caseína do leite, enquanto L. helveticus contém pelo menos duas dessas enzimas, a saber, PrtH e PrtH2 (Zhao et al., 2011). Assim, as altas proporções de sequências correspondentes às fortes espécies proteolíticas de L. helveticus e genes relacionados à proteólise podem se ligar ao conteúdo relativamente alto de peptídeos e aminoácidos livres em koumiss.
uma dificuldade na produção de koumiss em escala industrial é o controle da percepção do sabor, já que koumiss tem sido tradicionalmente feito por fermentação natural. Assim, tem sido difícil definir o sabor e o perfil dos principais componentes do sabor, particularmente na presença dos contaminantes naturais. A produção de componentes-chave de sabor é resultado da degradação fermentativa e enzimática de aminoácidos, como os aminoácidos de cadeia ramificada, metionina e aminoácidos aromáticos (Ardo, 2006). Exemplos de componentes de sabor incluem aldeídos, ácidos orgânicos e ésteres, que são formados por transaminase (AT)-pathway (Helinck et al., 2004). Esta via é iniciada por uma transaminase que catalisa a conversão de um aminoácido em seu α-ceto ácido correspondente (Helinck et al., 2004). Algumas sequências em nosso conjunto de dados corresponderam às putativas aminotransferases de cadeia ramificada e aminoácidos aromáticos. Particularmente, encontramos um aminoácido aromático putativo aminotransferase I no contig da espécie associada a koumiss, S. macedonicus. Como nosso trabalho atual anotou apenas o microbioma in silico, nossos dados não são suficientes para mostrar que essas sequências genéticas identificadas eram realmente funcionais para produzir os compostos de sabor koumiss mencionados. A presença desses genes, no entanto, sugere que eles são alguns possíveis candidatos para tais atividades fermentativas; no entanto, um trabalho experimental adicional seria necessário para elucidar seus papéis funcionais exatos.Além disso, encontramos sequências correspondentes a outras vias de conversão de aminoácidos, incluindo liases de aminoácidos e aldolase de treonina. A enzima anterior catalisa a conversão da metionina em metanetiol (Irmler et al., 2008), resultando assim em dimetil dissulfeto e dimetil trissulfeto formação (Fernandez et al., 2000), enquanto o último catalisa a conversão de treonina em glicina e acetaldeído (Ott et al., 2000). Da mesma forma, apenas localizar essas sequências dentro do microbioma koumiss não pode servir como evidência definitiva em seus papéis biológicos reais; futura confirmação experimental será necessária.
conclusão
o presente estudo utilizou um método metagenômico modificado para analisar o microbioma bacteriano de amostras de koumiss coletadas da Mongólia e mongólia interior da China. Caracterizamos os metagenomas filogênicos e funcionais das espécies raras em koumiss; e nosso conjunto de dados Reflete características que são de interesse e potencial da biotecnologia. Nosso estudo demonstrou pela primeira vez a viabilidade de incorporar as técnicas de amplificação unicelular na detecção da microbiota bacteriana de koumiss, bem como contaminantes microbianos. As técnicas aqui desenvolvidas podem ser utilizadas em estudos futuros para monitorar mudanças no microbioma de koumiss ao longo do processo de fermentação, com foco na população microbiana minoritária. Além disso, outras técnicas de omics, como transcriptômica, metabolômica, podem ser acopladas à análise metagenômica atual, a fim de confirmar as funções e a capacidade metabólica do microbioma koumiss.
tecnicamente, o próximo passo deste trabalho seria otimizar o método atual. Por exemplo, ao aumentar a diluição da amostra antes da amplificação do DNA, a chance de descobrir bactérias raras e novas pode ser melhorada. Por outro lado, uma técnica alternativa de sequenciamento que pode gerar leituras longas, como a única molécula de Biociências do Pacífico, a tecnologia de sequenciamento em tempo real, pode ser empregada para melhorar o processo de montagem do genoma.
contribuições do autor
WZ e HZ projetaram o estudo. WZ, GY, JY e L-YK escreveram o manuscrito. QH, WH, WL, BM E TS realizaram experimentos. WZ e QH analisaram dados. Todos os autores revisaram o manuscrito.
Declaração de conflito de interesses
os autores declaram que a pesquisa foi realizada na ausência de quaisquer relações comerciais ou financeiras que pudessem ser interpretadas como um potencial conflito de interesses.
reconhecimento
esta pesquisa foi apoiada pela Fundação Nacional de Ciências Naturais da China (concessão Nº 31571815).
Material Suplementar
O Material Suplementar para este artigo podem ser encontradas online em: https://www.frontiersin.org/article/10.3389/fmicb.2017.00165/full#supplementary-material
Altschul, S. F., Madden, T. L.; Schaffer, R. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST e PSI-BLAST: uma nova geração de programas de pesquisa de banco de dados de proteínas. Ácidos Nucleicos Res. 25, 3389-3402. doi: 10.1093/nar/25.17.3389
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Anastasiou, R., Driessche, G. V., Boutou, E., Kazou, M., Alexandraki, V., Vorgias, C. E., et al. (2015). Cepas projetadas de Streptococcus macedonicus em direção a um fenótipo osmótico resistente ao estresse mantêm sua capacidade de produzir a bacteriocina macedocina sob condições hiperosmóticas. J. Biotechnol. 212, 125–133. doi: 10.1016 / j. jbiotec.2015.08.018
Resumo PubMed / CrossRef Texto Completo / Google Scholar
Ardo, Y. (2006). Formação de sabor por catabolismo de aminoácidos. Biotechnol. Adv. 24, 238-242. doi: 10.1016 / j. biotechadv.2005.11.005
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Chen, Y. F., Zhao, W. J., Wu, R. N., Sol, Z. H., Zhang, W. Y. Wang, J. C., et al. (2014). Análise proteoma de Lactobacillus helveticus H9 durante o crescimento em leite desnatado. J. Dairy Sci. 97, 7413–7425. doi: 10.3168 / jds.2014-8520
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Dawson, K. L., Bell, A., Kawakami, R. P., Coley, K., Yates, G., & Collins, D. M. (2012). Transmissão de Mycobacterium orygis (M. tuberculosis complex species) de um paciente com tuberculose para uma vaca leiteira na Nova Zelândia. J. Clin. Microbiol. 50, 3136–3138. doi: 10.1128 / JCM.01652-12
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Doi, K., Mori, K., Mutaguchi, Y., Tashiro, K., Fujino, Y., Ohmori, T., et al. (2013). Sequência genômica preliminar do produtor de aminoácidos de cadeia ramificada D Lactobacillus otakiensis JCM 15040T, isolado de um picles Tradicional Japonês. Locutor Do Genoma. 1, e546-e513. doi: 10.1128 / genomeA.00546-13
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Fernandez, M., van Doesburg, W., Rutten, G. A., Marugg, J. D., Alting, A. C., van Kranenburg, R., et al. (2000). Análises moleculares e funcionais do gene metC de Lactococcus lactis, codificando cistationina beta-liase. Appl. Environ. Microbiol. 66, 42–48. doi: 10.1128 / AEM.66.1.42-48.2000
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Franciosi, E., Carafa, I., Nardin, T., Schiavon, S., Poznanski, E., Cavazza, A., et al. (2015). Biodiversidade e produção de ácido gama-aminobutírico por bactérias lácticas isoladas de queijos tradicionais de leite de vaca cru alpino. Biomed Res. Int. 2015, 625740. doi: 10.1155/2015/625740
Resumo do PubMed / texto completo do CrossRef / Google Scholar
Fu, L., Niu, B., Zhu, Z., Wu, S., e Li, W. (2012). CD-HIT: acelerado para agrupar os dados de sequenciamento de próxima geração. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
PubMed Abstract | CrossRef Full Text | Google Scholar
Georgalaki, M. D., Sarantinopoulos, P., Ferreira, E. S., De Vuyst, L., Kalantzopoulos, G., and Tsakalidou, E. (2000). Biochemical properties of Streptococcus macedonicus strains isolated from Greek Kasseri cheese. J. Appl. Microbiol. 88, 817–825. doi: 10.1046/j.1365-2672.2000.01055.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Gesudu, Q. M., Zheng, Y., Xi, X. X., Hou, Q. C., Xie, H. Y., Huang, W. Q., et al. (2016). Investigando a estrutura e dinâmica da população bacteriana no koumiss tradicional da Mongólia Interior Usando sequenciamento em tempo real de molécula única. J. Dairy Sci. 99, 7852–7863. doi: 10.3168 / jds.2016-11167
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Guarcello, R., Carpino, S., Gaglio, R., Pino, A., Rapisarda, T., Caggia, C., et al. (2016). Uma grande aplicação em escala de fábrica de bactérias autóctones de ácido láctico selecionadas para a produção de queijo DOP Pecorino Siciliano. Microbiol Alimentar. 59, 66–75. doi: 10.1016 / j. fm. 2016.05.011
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Hao, Y., Zhao, L., Zhang, H., Zhai, Z., Huang, Y., Liu, X., et al. (2010). Identificação da biodiversidade bacteriana em koumiss por eletroforese em gel gradiente desnaturante e reação em cadeia da polimerase específica da espécie. J. Dairy Sci. 93, 1926–1933. doi: 10.3168 / jds.2009-2822
PubMed Abstract / CrossRef texto completo / Google Scholar
Helinck, S., Le Bars, D., Moreau, D., and Yvon, M. (2004). Capacidade das bactérias termofílicas do ácido láctico para produzir compostos aromáticos a partir de aminoácidos. Appl. Environ. Microbiol. 70, 3855–3861. doi: 10.1128 / AEM.70.7.3855-3861.2004
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Hoskins, L. C., Agustines, M., McKee, W. B., Boulding, E. T., Kriaris, M., e Niedermeyer, G. (1985). Degradação da mucina nos ecossistemas do cólon humano. Isolamento e propriedades das cepas fecais que degradam antígenos do grupo sanguíneo ABH e oligossacarídeos das glicoproteínas da mucina. J. Clin. Investir. 75, 944–953. doi: 10.1172/JCI111795
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Irmler, S., Raboud, S., Beisert, B., Rauhut, D., e Berthoud, H. (2008). Clonagem e caracterização de dois genes Lactobacillus casei que codificam uma cistationina liase. Appl. Environ. Microbiol. 74, 99–106. doi: 10.1128 / AEM.00745-07
Resumo PubMed / CrossRef Texto Completo / Google Scholar
Jagielski, V. (1877). O valor de koumiss no tratamento de náuseas, vômitos e incapacidade de reter outros alimentos no estômago. Br. Med. J. 2, 919-921. doi: 10.1136 / bmj.2.887.919
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Kato, S., Ishihara, T., Hemmi, H., Kobayashi, H., e Yoshimura, T. (2011). Alterações nas concentrações de aminoácidos D e estruturas comunitárias microbianas durante a fermentação de vinhos tintos e brancos. J. Biosci. Bioeng. 111, 104–108. doi: 10.1016 / j. jbiosc.2010.08.019
resumo PubMed / CrossRef texto completo / Google Scholar
Li, H. e Durbin, R. (2009). Alinhamento rápido e preciso de Leitura curta com Burrows-Wheeler transform. Bioinformática 25, 1754-1760. doi: 10.1093/Bioinformática / btp324
resumo PubMed / CrossRef texto completo / Google Scholar
Li, W., e Godzik, A. (2006). CD-hit: um programa rápido para agrupar e comparar grandes conjuntos de sequências de proteínas ou nucleotídeos. Bioinformática 22, 1658-1659. doi: 10.1093/bioinformatics/btl158
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Miyamoto, M., Seto, T., Nakajima, H., Burenjargal, S., Gombojav, A., Demberei, S., et al. (2010). Análise de eletroforese em gel gradiente desnaturante de bactérias do ácido láctico e leveduras no leite fermentado mongol tradicional. Food Sci. Technol. Res. 16, 319-326. doi: 10.3136 / fstr.16.319
CrossRef texto completo / Google Scholar
Mulder, N. J., and Apweiler, R. (2008). O banco de dados InterPro e ferramentas para análise de domínio de proteínas. Moeda. Protoc. Bioinformática Capítulo 2, Unit2.7. doi: 10.1002 / 0471250953.bi0207s21
PubMed Abstract / CrossRef Full Text / Google Scholar
Noguchi, H., Park, J., e Takagi, T. (2006). MetaGene: gene procariótico que encontra das seqüências ambientais da espingarda do genoma Ácidos Nucleicos Res. 34, 5623-5630. doi: 10.1093/nar/gkl723
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A. A., Korobeynikov, A., Lapidus, A., et al. (2013). Montagem de genomas unicelulares e mini-metagenomas de produtos quiméricos MDA. J. Comput. Biol. 20, 714–737. doi: 10.1089 / cmb.2013.0084
PubMed Abstract / CrossRef texto completo / Google Scholar
Ott, A., Germond, J. E., E Chaintreau, A. (2000). Origem do acetaldeído durante a fermentação do leite usando (13) precursores rotulados em C. J. Agric. Comida Chem. 48, 1512–1517. doi: 10.1021 / jf9904867
PubMed Abstract / CrossRef Full Text / Google Scholar
Pan, D. D., Zeng, X. Q., e Yan, Y. T. (2011). Caracterização de Lactobacillus fermentum SM-7 isolado de koumiss, uma bactéria probiótica potencial com efeitos redutores de colesterol. J. Sci. Alimento. Agric. 91, 512–518. doi: 10.1002 / jsfa.4214
PubMed Abstract / CrossRef Texto Completo / Google Scholar
Papadimitriou, K., Anastasiou, R., Maistrou, E., Plakas, T., Papandreou, N. C., Hamodrakas, S. J., et al. (2015). Aquisição através da transferência horizontal de genes do plasmídeo pSMA198 por Streptococcus macedonicus ACA-DC 198 aponta para a origem leiteira da espécie. PLoS um 10: e0116337. doi: 10.1371 / jornal.pone.0116337
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Segata, N., Waldron, L., Ballarini, A., Narasimhan, V., Jousson, O., e Huttenhower, C. (2012). Perfil da comunidade microbiana metagenômica usando genes marcadores específicos do clado. Conversao. Métodos 9, 811-814. doi: 10.1038 / nmeth.2066
resumo PubMed / CrossRef texto completo / Google Scholar
Sun, Z., Liu, W., Zhang, J., Yu, J., Zhang, W., Cai, C., et al. (2010). Identificação e caracterização dos lactobacilos dominantes isolados de koumiss na China. J. Gen. Appl. Microbiol. 56, 257–265. doi: 10.2323 / jgam.56.257
PubMed Abstract / CrossRef Texto Completo / Google Scholar
Thompson, J. (1879). O valor do koumiss no desperdício de doenças. Br. Med. J. 1, 270. doi: 10.1136 / bmj.1.1466.270-C
PubMed Abstract / CrossRef texto completo / Google Scholar
Vincent, S. J., Faber, E. J., Neeser, J. R., Stingele, F., And Kamerling, J. P. (2001). Estrutura e propriedades do exopolissacarídeo produzido por Streptococcus macedonicus Sc136. Glicobiologia 11, 131-139. doi: 10.1093/glycob/11.2.131
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Wang, L., Christophersen, C. T., Sorich, M. J., Gerber, J. P., Angley, M. T., e Conlon, M. A. (2013). Aumento da abundância de Sutterella spp. e Ruminococcus torques em fezes de crianças com transtorno do espectro do autismo. Mol. Autismo 4, 42. doi: 10.1186/2040-2392-4-42
Resumo do PubMed / texto completo do CrossRef / Google Scholar
Ward, T. L., Hosid, S., Ioshikhes, I., e Altosaar, I. (2013). Metagenoma do leite humano: uma análise da capacidade funcional. BMC Microbiol. 13:116. doi: 10.1186/1471-2180-13-116
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Watanabe, K., Fujimoto, J., Tomii, Y., Sasamoto, M., Makino, H., Kudo, Y., et al. (2009). Lactobacillus kisonensis sp. novembro., Lactobacillus otakiensis sp. novembro., Lactobacillus rapi sp. novembro. e Lactobacillus sunkii sp. novembro., espécies heterofermentativas isoladas de sunki, um picles Japonês Tradicional. T. J. Syst. Evol. Microbiol. 59 (Pt 4), 754-760. doi: 10.1099 / ijs.0.004689-0
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Wu, R., Wang, L., Wang, J., Li, H., Menghe, B., Wu, J., et al. (2009). Isolamento e seleção probiótica preliminar de lactobacilos de koumiss na Mongólia Interior. J. Microbiol Básico. 49, 318–326. doi: 10.1002 / jobm.200800047
PubMed Abstract / CrossRef texto completo / Google Scholar
Zerbino, D. R., and Birney, E. (2008). Velvet: algoritmos para montagem de Leitura curta de novo usando gráficos de Bruijn. Genoma Res. 18, 821-829. doi: 10.1101 / gr.074492.107
resumo PubMed / CrossRef texto completo / Google Scholar
Zhang, W., Sun, Z., Sun, T., e, Zhang, H. (2010a). Triagem por PCR e análise de sequência de clusters de lio em cepas de Lactobacillus casei isoladas de koumiss. Folia Microbiol. (Praha) 55, 603-606. doi: 10.1007 / s12223-010-0097-3
Resumo do PubMed / texto completo do CrossRef / Google Scholar
Zhang, W., Yu, D., Sun, Z., Wu, R., Chen, X., Chen, W., et, al. (2010b). Sequência completa do genoma de Lactobacillus casei Zhang, uma nova cepa probiótica isolada do koumiss caseiro tradicional na Mongólia Interior, China. J. Bacteriol. 192, 5268–5269. doi: 10.1128 / JB.00802-10
resumo PubMed / CrossRef texto completo / Google Scholar
Zhang, W., e Zhang, H. (2011). “Fermentação e koumiss”, em Handbook of Food and Beverage Fermentation Technology, 2ª Edn, ed. Y. H. Hui (Boca Raton, FL: Crc Press), 165-172.
Google acadêmico
Zhang, W. Y., Chen, Y. F., Zhao, W. J., Kwok, L. Y., e Zhang, H. P. (2015). Expressão gênica do sistema proteolítico de Lactobacillus helveticus H9 durante a fermentação do leite. Anao. Microbiol. 65, 1171–1175. doi: 10.3168 / jds.2014-8520
PubMed Abstract / CrossRef texto completo / Google Scholar
Zhao, W., Chen, Y., Sun, Z., Wang, J., Zhou, Z., Sun, T., et, al. (2011). Sequência completa do genoma de Lactobacillus helveticus H10. J. Bacteriol. 193, 2666–2667. doi: 10.1128 / JB.00166-11
PubMed Abstract / CrossRef Texto Completo / Google Scholar