- wprowadzenie
- materiały i metody
- Przygotowanie próbek
- rozcieńczanie gradientu i Amplifikacja wielokrotnego przemieszczenia
- Budowa Biblioteki i sekwencjonowanie
- Analiza danych
- Kontrola jakości sekwencji i filtrowanie
- przypisanie taksonomiczne i różnorodność
- Czytaj Assembly, Gene Prediction, and Annotation
- numery akcesyjne sekwencji nukleotydów
- wyniki
- eksperymentalne Projektowanie i sekwencjonowanie
- Adnotacja taksonomiczna
- montaż Metagenomiczny, Przewidywanie genów i adnotacja funkcjonalna
- dyskusja
- wniosek
- autorzy
- Oświadczenie o konflikcie interesów
- Materiały uzupełniające
wprowadzenie
Koumiss, zwany także chige, chigo, arrag lub airag, w języku mongolskim, jest rodzajem tradycyjnego fermentowanego produktu mlecznego. Od wieków jest popularnym jedzeniem w Mongolii i Mongolii Wewnętrznej w Chinach (Zhang and Zhang, 2011). Mieszkańcy tych regionów spożywali kumiss podczas wielkich uroczystości i ofiarnych ofiar (Zhang and Zhang, 2011). Najwcześniejsze wzmianki o produkcji kumissu sięgają czasów dynastii Han (BC202-AD202). Produkt ten osiągnął szeroką popularność w okresie dynastii Yuan (AD1271-AD1368) (Zhang et al., 2010b). Obecnie kumiss jest powszechnym pokarmem dla miejscowej ludności Mongolii i Mongolii Wewnętrznej, chociaż tylko na kilku z tych obszarów jest produkowany na skalę przemysłową. Koumiss nie tylko dostarcza bogatych składników odżywczych, w tym wysokiej zawartości niezbędnych aminokwasów i witamin, ale uważa się, że łagodzi szeroki zakres schorzeń i jest korzystny dla opieki pooperacyjnej (Jagielski, 1877; Thompson, 1879).
tradycyjnie koumiss jest zwykle produkowany w drewnianych beczkach, pojemnikach ze skóry zwierzęcej lub urnach. Fermentacja zachodzi naturalnie w temperaturze otoczenia po dodaniu przesączonego mleka klaczy do pojemnika ze starym kumysem, który służy jako kultura starterowa (Zhang and Zhang, 2011). Koumiss jest dobrym źródłem nowych bakterii o potencjale biotechnologicznym (Zhang et al., 2010a; Pan et al., 2011). Dlatego bardzo interesujące jest zbadanie i zachowanie jak największej liczby bakterii koumiss związanych z fermentacją. W ciągu ostatnich dziesięcioleci przeprowadzono szereg badań w celu zbadania społeczności bakterii koumiss (Wu et al., 2009; Hao et al., 2010), głównie badane metodami opartymi na kulturze, biologii molekularnej i pirosekwencji (Sun et al., 2010). Wśród tych różnych podejść, metoda oparta na pyrosekwencjonowaniu zapewniła najbardziej wszechstronny profil mikrobioty kumissa, niezależny od cech fenotypowych i problemów z uprawnością poszczególnych drobnoustrojów. Jednak spektrum funkcjonalnych genów kodowanych przez bakterie kumiss i ich zdolności fermentacyjne pozostaje słabo scharakteryzowane, szczególnie w rzadkich populacjach drobnoustrojów.
w niniejszym badaniu wykorzystano technikę genomiki jednokomórkowej do analizy bakteryjnych metagenomów 10 próbek kumissa pobranych z Mongolii i Mongolii Wewnętrznej w Chinach. Obecne prace wykorzystały najnowocześniejsze technologie w badaniu różnorodności bakterii w produktach mlecznych. Nasze prace wykazały wykonalność odkrycia taksonów o niskiej obfitości poprzez zastosowanie podejścia metagenomiki jednokomórkowej. Zachęcające wyniki promowałyby rozwój i zastosowanie nowatorskich podejść w rozwiązywaniu problemów w tradycyjnej dziedzinie badań.
materiały i metody
Przygotowanie próbek
łącznie pobrano 10 próbek kumissa z Mongolii (MG14, MG15, MG16, MG17 i MG18) i Mongolii Wewnętrznej w Chinach (NM17, NM18, NM19, NM20 i NM21) do badania metagenomiki. Próbki pobierano w warunkach aseptycznych i transportowano w suchym lodzie.
jeden mililitr każdej próbki został wstępnie przetworzony zgodnie z metodologią opisaną w Ward et al. (2013) z pewnymi modyfikacjami. Krótko, próbki rozmrażano w łaźni lodowej przez 3-5 minut. Po stopieniu próbek poddano je wirowaniu z małą prędkością w celu usunięcia zanieczyszczeń i grudek komórek eukariotycznych. Komórki prokariotyczne następnie granulowano z surowic mlecznych przez odwirowanie w temperaturze 13 000 × g przez 15 minut. Granulki ponownie zawieszono w 2 mL soli fizjologicznej buforowanej fosforanem (PBS) z 1% Tritonem X-100 i inkubowano przez 2 godziny w temperaturze 37°C w celu lizy pozostałych komórek eukariotycznych. Następnie bakterie granulowano przez odwirowanie w temperaturze 13 000 × g przez 15 minut i granulki ponownie zawieszono w 500 µL PBS. Na koniec, etap wirowania powtórzono jeszcze raz, aby umyć komórki bakteryjne.
rozcieńczanie gradientu i Amplifikacja wielokrotnego przemieszczenia
w celu wykrycia bakterii o niskiej obfitości, zawiesinę bakteryjną otrzymaną z każdej próbki kumissa rozcieńczano seryjnie w celu późniejszej reakcji amplifikacji. Liczba komórek w każdej próbce została z grubsza oszacowana pod mikroskopem (Nikon, Tokio, Japonia) za pomocą komory liczenia komórek (Qiujing, Szanghaj, Chiny). Etap rozcieńczania był kontynuowany, aż liczba komórek w każdej zawiesinie bakteryjnej osiągnie około 100. Amplifikację wielokrotnego przemieszczenia rozcieńczonych komórek przeprowadzono przy użyciu zestawu REPLI-G Single Cell Kit (Qiagen, Germantown, MD, USA) zgodnie z instrukcjami producenta.
Budowa Biblioteki i sekwencjonowanie
amplifikowane DNA ścięto losowo i wybrano fragmenty o długości około 500 bp. Po wybudowaniu biblioteki do kontroli jakości Biblioteki wykorzystano PerkinElmer LabChip® GX Touch oraz system PCR w czasie rzeczywistym StepOnePlusTM. Ostatecznie, zgodnie z instrukcjami producenta, na platformie Illumina HiSeq 2500 zsekwencjonowano sparowane odczyty 125-bp.
Analiza danych
Kontrola jakości sekwencji i filtrowanie
odczyty surowe generowane przez sekwencer mogą zawierać sztuczne odczyty skażenia adaptera podczas budowy biblioteki. Dlatego przeprowadzono trzy kroki w celu uzyskania wysokiej jakości czystego zestawu danych odczytu: (1) eliminacja odczytów spowodowanych zanieczyszczeniem adaptera; (2) Usunięcie odczytów ze średnim wynikiem poniżej wyniku phred Q30, który został uznany za najniższy odcięcia dla wysokiej jakości Bazy; (3) usunięcie odczytów ze znacznym nadmiarem „N” (≥5% odczytu). Analiza dalszych działań została oparta na czystych danych. Ponadto obliczono jakość bazy statystycznej w oparciu o Q30 i zawartość GC.
dopasowania do genomu gospodarza przeprowadzono w celu usunięcia sekwencji zanieczyszczeń pochodzących z gospodarza. Wszelkie odczyty pochodzące od gospodarza odrzucono przed dalszym porównaniem z sekwencjami referencyjnymi genomu bakterii (lub wirusów). Aby uzyskać dokładniejsze wyniki, w Ustawieniach użyto Modelu Mem Burrowsa–Wheelera (BWA) (Wersja 0.97 a) (Li i Durbin, 2009).
przypisanie taksonomiczne i różnorodność
oprogramowanie internetowe Metaflan zostało użyte do przypisania taksonomicznego do poziomu rodzaju i gatunku (Segata et al., 2012). Aby porównać różnorodność gatunków wewnątrz i między próbkami, przeanalizowaliśmy różnorodność alfa i beta za pomocą pakietu związanego z R.
Czytaj Assembly, Gene Prediction, and Annotation
aby uzyskać bardziej wyczerpujące informacje, zmontowaliśmy ścięte fragmenty w Genom (contigs). Jednak ze względu na obecność wielu gatunków, co jest powszechne w próbkach metagenomicznych, ulepszyliśmy bioinformatyczną metodę montażu genomu zwykle stosowaną do analizy pojedynczych gatunków poprzez integrację Pik (Wersja 3.6.2), skryptów wewnętrznych i metagenomicznych baz danych (Zerbino and Birney, 2008; Nurk et al., 2013).
oprogramowanie MetaGeneMark zostało wykorzystane do przewidywania genów zmontowanych stygów (Noguchi et al., 2006). Zbędne geny zostały usunięte za pomocą CD-HIT z pokryciem 90 I 95% tożsamości (Li i Godzik, 2006; Fu et al., 2012). Względna obfitość genów została określona przez dostosowanie wysokiej jakości sekwencjonowania do katalogu genów przy użyciu tej samej procedury. Późniejsza analiza rozbieżności opierała się na względnej obfitości genu. Adnotację genów przeprowadzono poprzez dopasowanie wysokiej jakości sekwencji do kilku publicznych baz danych (np. NCBI non-redundant database, NR; Clusters of Orthologous Groups of proteins, COGs; Kyoto Encyclopedia of Genes and Genomes, KEGG) za pomocą BLAST (Altschul et al., 1997). Wyszukiwanie domen zostało przeprowadzone za pomocą Interproscan (Mulder and Apweiler, 2008).
numery akcesyjne sekwencji nukleotydów
dane dotyczące sekwencji zgłoszone w tym badaniu zostały zdeponowane w bazie danych SRA (nr akcesyjny.SRP083102).
wyniki
eksperymentalne Projektowanie i sekwencjonowanie
aby wykryć nisko obfite bakterie w kumiss, zastosowano technikę amplifikacji jednokomórkowej do analizy metagenomów próbek. Trzy zawiesiny bakterii, każda po około 100 komórek, uzyskano z niezależnej próbki kumissa poprzez seryjne rozcieńczenie. Przeanalizowano łącznie 30 rozcieńczonych zawiesin. Każda rozcieńczona próbka otrzymała inny kod próbki, tj. numer identyfikacyjny próbki, po którym następowały 1, 2 lub 3, reprezentujące trzy oddzielne rozcieńczenia. Opierając się na założeniu, że niektóre rzadkie gatunki będą obecne w jednym z rozcieńczeń, przeprowadzono wielokrotne amplifikacje przemieszczeń komórek; i wygenerowano około 5 Gb danych dla każdej zawiesiny bakterii koumiss.
łącznie z 10 próbek koumissa wygenerowano 1 040 323 864 odczytów surowych (łącznie 30 zawiesin bakteryjnych). Średnia liczba odczytów dla każdego 100-komórkowego zawieszenia wynosiła 34 677 462 (tabela uzupełniająca S1). Po przycięciu i filtrowaniu sekwencji niekwalifikowanych uzyskaliśmy 1 018 381 702 czystych odczytów dla wszystkich próbek (tabela uzupełniająca S1). Wartości indeksu Shannona, indeksu Simpsona, indeksu Chao1 i liczby obserwowanych gatunków (rys. 1-4) wykazały, że większość próbek koumissa ma wysoką bioróżnorodność bakterii. Krzywe różnorodności Shannona-Wienera wykazały, że głębokość sekwencji jest odpowiednia dla wszystkich próbek (ryc. 1).
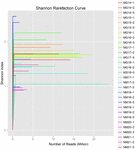
RYSUNEK 1. Krzywe rarefaction Shannona, które szacują różnorodność mikrobiologiczną próbek kumissa.
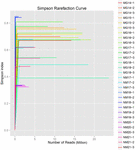
RYSUNEK 2. Krzywe Simpsona, które szacują różnorodność mikrobiologiczną próbek kumissa.
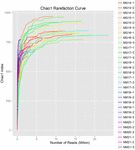
RYSUNEK 3. Chao1 krzywe rozrzedzenia, które szacują różnorodność mikrobiologiczną próbek kumissa.
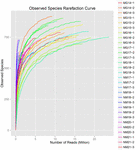
RYSUNEK 4. Obserwowany wskaźnik rozrzedzenia gatunków, który szacuje różnorodność mikrobiologiczną próbek kumissa.
Adnotacja taksonomiczna
wysokiej jakości sekwencje zostały przypisane do różnych poziomów taksonomicznych, aby umożliwić dogłębną analizę próbek społeczności bakteryjnych. W odniesieniu do niektórych opublikowanych badań nad bioróżnorodnością kumissa (Wu et al., 2009; Hao et al., 2010; Sun et al., 2010), sklasyfikowaliśmy znane i wcześniej nie zgłaszane bakterie związane z koumiss jako taksony, odpowiednio, pospolite i rzadkie.
sekwencje wysokiej jakości reprezentowały 13 różnych rodzajów (rycina 5). Trzy z nich miały średnią względną obfitość ponad 1%, w tym Lactobacillus (L.), Lactococcus i Streptococcus. Szczególnie Lactobacillus i Lactococcus były dwoma najliczniejszymi rodzajami występującymi w kumiss. Odsetek bakterii Lactobacillus w próbkach wynosił od 52,72 do 99,96%. Dwóch przedstawicieli tego rodzaju, L. helveticus i L. kefiranofaciens, było dominowanych wśród większości próbek kumissa (ryc. 6). Gatunek L. buchneri był obecny głównie w próbkach mongolskich, podczas gdy Lactococcus lactis był wykrywany w większości próbek niezależnie od regionu pobierania próbek.
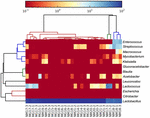
RYSUNEK 5. Heatmapa pokazująca względną obfitość bakterii wykrytych w próbkach kumissa na poziomie rodzaju.
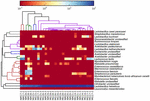
RYSUNEK 6. Heatmapa pokazująca względną obfitość bakterii wykrytych w próbkach kumissa na poziomie gatunku.
montaż Metagenomiczny, Przewidywanie genów i adnotacja funkcjonalna
montaż odczytów spowodował Długość zespołu 614 392 623 bp. Wartości N50 dla zespołów wynosiły od 5596 do 35200 bp (tabela uzupełniająca S2). Liczba przewidywanych genów w próbkach kumissa wahała się od 10 347 do 34 547, a średnia długość wynosiła od 647,82 do 985,33 bp (tabela uzupełniająca S3). Chociaż empiryczne analizy funkcjonalne genów wykraczały poza zakres obecnego badania, opisaliśmy mikrobiomy bakterii koumiss przy użyciu baz danych COG i KEGG, które przewidywały funkcję genów w dużej mierze w oparciu o homologię sekwencji.
łącznie 545 dopasowań w wyniku adnotacji wykazało wysoką homologię genów wykorzystania laktozy (Kategoria COGs węglowodanów i metabolizmu, G), a niektóre z nich znajdowały się w obrębie tego samego contig (Tabela 1).
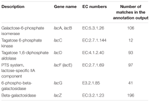
TABELA 1. Geny związane z metabolizmem laktozy zostały opisane w metagenomie bakterii kumiss.
niektóre inne sekwencje mogą kodować domniemane geny w kategorii COGs transportu i metabolizmu aminokwasów (E), w tym sekwencje odpowiadające białkom rozkładającym kazeinę, System Opp do pobierania oligopeptydów o 4-18 reszt i aminopeptydazy (np. iminopeptydazy i endopeptydazy). Chociaż niektóre sekwencje wykazywały duże podobieństwa z aminotransferazami specyficznymi dla argininy, asparaginianu, metioniny i izoleucyny, zidentyfikowano tylko jedną istotną zgodność, która odpowiadała przypuszczalnej domenie S. macedonicus-pochodzącej z klasy I/klasy II (IPR004839)-zawierającej aminotransferazę specyficzną dla tyrozyny i fenyloalaniny. Wreszcie, wiele sekwencji dzieliło wysoką homologię z liazami aminokwasowymi, w tym liazą s-rybosylhomocysteiny, liazą argininobursztynianową, asparaginianową liazą amoniakalną, gamma-liazą cystationinową, histydynową liazą amoniakalną i o-acetylohomoseryną (tiolową).
dyskusja
Koumiss jest popularnym tradycyjnym fermentowanym produktem mlecznym w Mongolii i Mongolii Wewnętrznej w Chinach. Chociaż przeprowadzono szereg badań w celu zbadania różnorodności bakterii w koumiss, uzyskano niewiele informacji na temat genetycznej zdolności mikrobioty koumiss. Tutaj zastosowaliśmy technikę amplifikacji jednokomórkowej do analizy metagenomów bakterii koumiss, szczególnie skupiając się na populacji bakterii o niskiej obfitości.
typowe podejście metagenomiczne zostało wcześniej zastosowane do opisu mikrobioty kumissa. Jednak ze względu na wysokie koszty uzyskania głębokiej sekwencji, rzadka populacja mikrobioty w próbkach jest często niedostatecznie pokryta. Tak więc zarówno filogenetyczne, jak i funkcjonalne metagenomy bakterii mniejszościowych pozostają ograniczone. Nasza metoda amplifikacji jednokomórkowej polegała na seryjnym rozcieńczeniu próbek do 100-komorowych zawiesin. Ze względu na małą liczbę komórek obecnych w rozcieńczonych próbkach kumissa, wyodrębniono jedynie ograniczoną ilość DNA. Etap amplifikacji zwiększył ilość materiałów DNA, które mają być analizowane, a tym samym ułatwił analizę metagenomu próbek zawierających niewielkie ilości DNA. Jedną z wad tej metody była trudność w zapewnieniu, że sekwencje każdego taksonu będą jednakowo amplifikowane w procesie; w związku z tym uzyskane tutaj wyniki mogą odzwierciedlać tylko względną obfitość sekwencji, ale nie bezwzględne ilości zidentyfikowanych taksonów lub funkcjonalnych genów. Jednak nie miałoby to żadnego wpływu na naszą analizę, ponieważ niniejsze badanie różni się od innych opublikowanych prac skupianiem się na rzadkiej populacji bakterii. Dane uzyskane w wyniku tej pracy dostarczają informacji uzupełniających niedostatecznie reprezentowanej populacji. Uważamy, że obecne podejście jest odpowiednie do przyszłej analizy różnorodności bakterii dla różnych rodzajów środowisk ekologicznych.
mikrobiota bakteryjna kumissa składa się głównie z bakterii kwasu mlekowego (LAB) i niektórych bakterii kwasu octowego (Zhang and Zhang, 2011). Zgodnie z oczekiwaniami, nasz zbiór danych zawierał głównie sekwencje reprezentujące bakterie laboratoryjne i kwas octowy. Sekwencje odpowiadające Lactobacillus helveticus dominowały we wszystkich próbkach. Poza tym sekwencje reprezentujące gatunek „L. kefiranofaciens”. L. buchneri. L. kefiranofaciens. Enterococcus (E.) casseliflavus. E. faecalis. E. faecium. Leuconostoc mesenteroides. Stwierdzono również Lactococcus lactis i Acetobacter pasteurianus. Identyfikacja sekwencji różnych taksonów może nie wystarczyć do wykazania żywotności bakterii, niemniej jednak odzwierciedla społeczność bakteryjną w pewnym momencie procesu fermentacji. Miyamoto et al. (2010) sugeruje, że występowanie bakterii w końcowych produktach fermentowanych jest związane z ich tolerancją na stres kwasowy. Ogólnie rzecz biorąc, lactobacilli mają wyższą tolerancję na kwasy niż laktokoki, co może wyjaśniać wysoką względną obfitość sekwencji lactobacilli obecnych w naszym zbiorze danych. Z drugiej strony, częste występowanie L. helveticus w kumiss zbiegł się z obserwacją dominacji sekwencji L. helveticus.
w naszym zbiorze danych niektóre sekwencje odpowiadały L. otakiensis, która jest rzadkim gatunkiem, który nigdy nie został zgłoszony w kumiss lub innych niszach mlecznych. Gatunek został po raz pierwszy opisany i wyizolowany z nieosolonego roztworu marynowanego stosowanego w produkcji sunki, tradycyjnego japońskiego marynatu (Watanabe et al., 2009). Został odkryty przez profilowanie polimorfizmu o amplifikowanej długości fragmentu w oparciu o Gen recA. Od tego czasu gatunek ten nie był kojarzony z innymi niszami związanymi z pożywieniem. Tak więc jest prawdopodobne,że ta bakteria należy do autochtonicznej flory ogórków. Nie można jednak wykluczyć, że nie został on znaleziony tylko ze względu na czułość zastosowanej metody wykrywania. Lactobacillus otakiensis może wytwarzać aminokwasy rozgałęzione D, takie jak D-leucyna, d-allo-izoleucyna i D-walina (Doi i in., 2013). Ma potencjał do wykorzystania w poprawie właściwości produkcyjnych niektórych sfermentowanych produktów spożywczych (Kato et al., 2011).
nasze badania zidentyfikowały również sekwencje reprezentujące gatunek S. macedonicus, który nigdy nie został zgłoszony jako bakteria związana z kumissem. Zamiast tego jest to kultura starterowa obecna w greckich serach Owczych i kozich (Georgalaki et al., 2000). Niektórzy przedstawiciele tego gatunku są w stanie wytwarzać egzopolisacharydy, bakteriocyny (Vincent i wsp., 2001; Anastasiou et al., 2015) oraz kwas gamma-aminomasłowy (Franciosi et al., 2015). Chociaż gatunek ten jest często izolowany ze sfermentowanej żywności, pierwotna nisza S. macedonicus była kontrowersyjna (Guarcello et al., 2016). Nie do niedawna, Papadimitriou et al. (2015) zidentyfikował nabyty plazmid, pSMA198, z Lactococcus lactis. Plazmid był prawdopodobnie przenoszony przez zdarzenie wymiany genetycznej przodków w środowisku produktów mlecznych, wskazując na pochodzenie mleczne S. macedonicus(Papadimitriou et al., 2015). Podobny do S. thermophilus. S. macedonicus jest blisko spokrewniony z komensalami i patogenami oportunistycznymi kompleksu S. bovis/S. equinus.
identyfikacja sekwencji reprezentujących gatunek Ruminococcus torques była nieoczekiwana, ponieważ bakteria ta zwykle znajduje się w środowisku jelitowym. Jest to normalny mikrob jelitowy człowieka, który może degradować oligosacharydy mucyny przez konstytutywną produkcję wydzielniczych glikozydaz(Hoskins et al., 1985). Ostatnie dowody kliniczne pokazują, że obfitość kału tego gatunku zmienia się u dzieci z zaburzeniami ze spektrum autyzmu; jednak jego rola w zaburzeniu pozostaje niejasna(Wang et al., 2013).
co zaskakujące, niektóre sekwencje reprezentowały potencjalne patogeny. Na przykład zapalenie płuc Klebsiella jest oportunistycznym patogenem ludzkim, który zamieszkuje około 40% jelit ludzkich i zwierzęcych. Mycobacterium orygis, wcześniej nazywany Bacillus oryx, jest członkiem kompleksu Mycobacterium tuberculosis, który może powodować ludzką gruźlicę (Dawson i wsp ., 2012). Te dwa gatunki odnotowano w mleku surowym, ale nie kumysie. W związku z tym ich obecność może być przypisana zanieczyszczeniu podczas konwencjonalnej produkcji kumissa, zwłaszcza w Warunkach manipulacji bez lub w warunkach niskiej aseptyki.
metabolizm bakterii odgrywa ważną rolę w kształtowaniu cech i jakości koumiss. Procesy mikrobiologiczne, takie jak lipoliza i proteoliza, są wymagane do syntezy związków aromatycznych i smakowych kumissa (Gesudu et al., 2016). Konsekwentnie, bakteryjny metagenom zawierał sekwencje, które potencjalnie kodują degradację laktozy i systemy proteolityczne. W przeciwieństwie do stosunkowo prostych szlaków katabolicznych laktozy, systemy proteolityczne laboratorium składają się z różnorodnych enzymów (Chen et al., 2014). W porównaniu z innymi wykrytymi koumiss LAB, dominujący gatunek L. helveticus charakteryzuje się wysoką aktywnością proteolityczną (Zhang et al., 2015). Większość laboratoriów posiada tylko jedną proteinazę otoczki komórkowej, która inicjuje hydrolizę kazeiny mlecznej, podczas gdy L. helveticus zawiera co najmniej dwa z tych enzymów, a mianowicie PrtH i PrtH2 (Zhao et al., 2011). Tak więc, wysokie proporcje sekwencji odpowiadających silnym gatunkom proteolitycznym L. helveticus i Genom związanym z proteolizą mogą wiązać się ze stosunkowo wysoką zawartością peptydów i wolnych aminokwasów w kumiss.
jedną z trudności w produkcji koumissa na skalę przemysłową jest kontrola percepcji smaku, ponieważ koumiss był tradycyjnie wytwarzany przez naturalną fermentację. Tak więc trudno było zdefiniować smak i profil kluczowych składników smakowych, szczególnie w obecności naturalnych zanieczyszczeń. Produkcja kluczowych składników smakowych jest wynikiem fermentacyjnej i enzymatycznej degradacji aminokwasów, takich jak aminokwasy rozgałęzione, metionina i aminokwasy aromatyczne (Ardo, 2006). Przykłady składników smakowych obejmują aldehydy, kwasy organiczne i estry, które są tworzone przez transaminazę (at) – szlak (Helinck et al., 2004). Szlak ten jest inicjowany przez transaminazę, która katalizuje konwersję aminokwasu do odpowiadającego mu kwasu α-ketonowego(Helinck i in., 2004). Niektóre sekwencje w naszym zbiorze danych odpowiadały przypuszczalnym aminotransferazom aminokwasów rozgałęzionych i aromatycznych. W szczególności znaleźliśmy przypuszczalny aminokwas aromatyczny aminotransferazę I w contig gatunku S. macedonicus związanego z kumissem. Ponieważ nasza obecna praca opisywała tylko mikrobiom in silico, nasze dane nie są wystarczające, aby pokazać, że te zidentyfikowane sekwencje genów były rzeczywiście funkcjonalne do produkcji wyżej wymienionych związków smakowych kumissa. Obecność tych genów sugeruje jednak, że są one niektórymi możliwymi kandydatami do takich działań fermentacyjnych; jednak dalsze prace eksperymentalne byłyby wymagane, aby wyjaśnić ich dokładną rolę funkcjonalną.
co więcej, znaleźliśmy sekwencje odpowiadające innym szlakom konwersji aminokwasów, w tym liazom aminokwasowym i aldolazie treoninowej. Ten pierwszy enzym katalizuje konwersję metioniny do metanotiolu (Irmler i wsp ., 2008), w wyniku czego powstaje dwusiarczek dimetylu i trisiarczek dimetylu (Fernandez et al., 2000), podczas gdy ten ostatni katalizuje konwersję treoniny do glicyny i aldehydu octowego (Ott et al., 2000). Podobnie samo zlokalizowanie tych sekwencji w mikrobiomie kumissa nie może służyć jako konkretny dowód w ich rzeczywistych rolach biologicznych; wymagane będzie przyszłe potwierdzenie eksperymentalne.
wniosek
w niniejszym badaniu wykorzystano zmodyfikowaną metodę metagenomiki do analizy mikrobiomu bakteryjnego próbek kumissa pobranych z Mongolii i Mongolii Wewnętrznej w Chinach. Scharakteryzowaliśmy zarówno filogenetyczne, jak i funkcjonalne metagenomy rzadkich gatunków w kumiss; a nasz zbiór danych odzwierciedla cechy, które są interesujące i potencjalne w biotechnologii. Nasze badania wykazały po raz pierwszy możliwość zastosowania technik amplifikacji jednokomórkowej w wykrywaniu mikrobioty bakteryjnej kumissa, a także zanieczyszczeń mikrobiologicznych. Techniki opracowane w niniejszym dokumencie mogą być wykorzystane w przyszłych badaniach w celu monitorowania zmian w mikrobiomie kumissa wzdłuż procesu fermentacji, ze szczególnym uwzględnieniem mniejszościowej populacji drobnoustrojów. Ponadto, inne techniki omics, tak jak transkryptomics, metabolomics, mogą łączyć się z aktualną metagenomics analizą w celu potwierdzenia funkcjami i metabolicznymi zdolnościami koumiss microbiome.
technicznie następnym krokiem tej pracy byłoby zoptymalizowanie obecnej metody. Na przykład, zwiększając rozcieńczenie próbki przed amplifikacją DNA, można zwiększyć szansę na odkrycie rzadkich i nowych bakterii. Z drugiej strony, alternatywny sekwencjonowanie technika który móc długi czyta, tak jak Pacific Biosciences single molekuła, real-time sekwencjonowanie technologia, móc zatrudniać ulepszać Genom assembling proces.
autorzy
WZ i HZ zaprojektowali opracowanie. Rękopis został napisany przez WZ, GY, JY i L-YK. QH, WH, WL, BM i TS przeprowadzili eksperymenty. WZ i QH analizowały dane. Wszyscy autorzy recenzowali manuskrypt.
Oświadczenie o konflikcie interesów
autorzy oświadczają, że badanie zostało przeprowadzone przy braku jakichkolwiek relacji handlowych lub finansowych, które mogłyby być interpretowane jako potencjalny konflikt interesów.
badania te były wspierane przez National Natural Science Foundation Of China (Grant No.31571815).
Materiały uzupełniające
Materiały uzupełniające do tego artykułu można znaleźć w Internecie pod adresem: https://www.frontiersin.org/article/10.3389/fmicb.2017.00165/full#supplementary-material
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST i PSI-BLAST: nowa generacja programów do wyszukiwania baz danych białek. Nucleic Acids Res. 25, 3389-3402. doi: 10.1093/nar / 25.17.3389
PubMed Abstract / CrossRef Full Text / Google Scholar
Anastasiou, R., Driessche, G. V., Boutou, E., Kazou, M., Alexandraki, V., Vorgias, C. E., et al. (2015). Zmodyfikowane szczepy Streptococcus macedonicus w kierunku fenotypu odpornego na stres osmotyczny zachowują zdolność do produkcji bakteriocyny macedocyny w Warunkach hiperosmotycznych. J. Biotechnol. 212, 125–133. doi: 10.1016 / j.jbiotec.2015.08.018
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
Ardo, Y. (2006). Tworzenie smaku przez katabolizm aminokwasów. Biotechnol. ADV. 24, 238-242. doi: 10.1016 / j.2005.11.005
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Chen, Y. F., Zhao, W. J., Wu, R. N., Sun, Z. H., Zhang, W. Y., Wang, J. C., et al. (2014). Analiza proteomu Lactobacillus helveticus H9 podczas wzrostu mleka odtłuszczonego. J. Dairy Sci. 97, 7413–7425. doi: 10.3168 / jds.2014-8520
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Dawson, K. L., Bell, A., Kawakami, R. P., Coley, K., Yates, G., and Collins, D. M. (2012). Przeniesienie Mycobacterium orygis (M. tuberculosis Complex species) z chorego na gruźlicę na krowę mleczną w Nowej Zelandii. J. Clin. Mikrobiol. 50, 3136–3138. doi: 10.1128 / JCM.01652-12
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
doi, K., Mori, K., Mutaguchi, Y., Tashiro, K., Fujino, Y., Ohmori, T., et al. (2013). Projekt sekwencji genomu producenta aminokwasów rozgałęzionych D-Lactobacillus otakiensis JCM 15040T, wyizolowanego z tradycyjnego japońskiego ogórka. Ogłoszenie Genomu. 1, e546-e513. doi: 10.1128 / genomeA.00546-13
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Fernandez, M., van Doesburg, W., Rutten, G. A., Marugg, J. D., Alting, A. C., van Kranenburg, R., et al. (2000). Molekularne i funkcjonalne analizy genu metC Lactococcus lactis kodującego beta-liazę cystationiny. Appl. Environ. Mikrobiol. 66, 42–48. doi: 10.1128/AEM.66.1.42-48.2000
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Franciosi, E., Carafa, I., Nardin, T., Schiavon, S., Poznański, E., Cavazza, A., et al. (2015). Bioróżnorodność i produkcja kwasu gamma-aminomasłowego przez bakterie kwasu mlekowego wyizolowane z tradycyjnych alpejskich serów surowego mleka krowiego. Biomed Res.Int. 2015, 625740. doi: 10.1155/2015/625740
PubMed Abstract / CrossRef Pełny tekst / Google Scholar
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: akcelerator do klastrowania sekwencjonowania danych nowej generacji. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
PubMed Abstract | CrossRef Full Text | Google Scholar
Georgalaki, M. D., Sarantinopoulos, P., Ferreira, E. S., De Vuyst, L., Kalantzopoulos, G., and Tsakalidou, E. (2000). Biochemical properties of Streptococcus macedonicus strains isolated from Greek Kasseri cheese. J. Appl. Microbiol. 88, 817–825. doi: 10.1046/j.1365-2672.2000.01055.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Gesudu, Q. M., Zheng, Y., Xi, X. X., Hou, Q. C., Xie, H. Y., Huang, W. Q., et al. (2016). Badanie struktury i dynamiki populacji bakterii w tradycyjnym kumisie z Mongolii Wewnętrznej przy użyciu sekwencjonowania w czasie rzeczywistym pojedynczej cząsteczki. J. Dairy Sci. 99, 7852–7863. doi: 10.3168 / jds.2016-11167
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Guarcello, R., Carpino, S., Gaglio, R., Pino, A., Rapisarda, T., Caggia, C., et al. (2016). Duże fabryczne zastosowanie wybranych autochtonicznych bakterii kwasu mlekowego do produkcji sera PDO Pecorino Siciliano. Mikrobiol Spożywczy. 59, 66–75. 10.1016 / j. fm. 2016.05.011
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Hao, Y., Zhao, L., Zhang, H., Zhai, Z., Huang, Y., Liu, X., et al. (2010). Identyfikacja bioróżnorodności bakterii w kumissie poprzez denaturowanie elektroforezy w żelu gradientowym i specyficznej dla gatunku reakcji łańcuchowej polimerazy. J. Dairy Sci. 93, 1926–1933. doi: 10.3168 / jds.2009-2822
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Helinck, S., Le Bars, D., Moreau, D., and Yvon, M. (2004). Zdolność termofilnych bakterii kwasu mlekowego do wytwarzania związków aromatycznych z aminokwasów. Appl. Environ. Mikrobiol. 70, 3855–3861. doi: 10.1128/AEM.70.7.3855-3861.2004
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Hoskins, L. C., Agustines, M., McKee, W. B., Boulding, E. T., Kriaris, M., and Niedermeyer, G. (1985). Degradacja mucyny w ekosystemach jelita grubego człowieka. Izolacja i właściwości szczepów kału, które degradują antygeny grupy krwi ABH i oligosacharydy z glikoprotein mucyny. J. Clin. Inwestuj. 75, 944–953. doi: 10.1172 / JCI111795
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Irmler, S., Raboud, S., Beisert, B., Rauhut, D., and Berthoud, H. (2008). Klonowanie i charakterystyka dwóch genów Lactobacillus casei kodujących liazę cystationinową. Appl. Environ. Mikrobiol. 74, 99–106. doi: 10.1128/AEM.00745-07
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
Jagielski, V. (1877). Wartość kumiss w leczeniu nudności, wymiotów i niemożności utrzymania innych pokarmów na żołądku. Br. Med. J. 2, 919-921. doi: 10.1136 / bmj.2.887.919
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Kato, S., Ishihara, T., Hemmi, H., Kobayashi, H., and Yoshimura, T. (2011). Zmiany w stężeniach D-aminokwasów i strukturach wspólnoty mikrobiologicznej podczas fermentacji win czerwonych i białych. J. Biosci. Bioeng. 111, 104–108. doi: 10.1016 / j.jbiosc.2010.08.019
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Li, H., and Durbin, R. (2009). Szybkie i dokładne dopasowanie do krótkiego odczytu z transformatą Burrowsa-Wheelera. Bioinformatyka 25, 1754-1760. doi: 10.1093/bioinformatics / btp324
PubMed Abstract / CrossRef Full Text / Google Scholar
Li, W., and Godzik, A. (2006). Cd-hit: szybki program do grupowania i porównywania dużych zestawów sekwencji białek lub nukleotydów. Bioinformatyka 22, 1658-1659. doi: 10.1093/bioinformatics / btl158
PubMed Abstract / CrossRef Full Text / Google Scholar
Miyamoto, M., Seto, T., Nakajima, H., Burenjargal, S., Gombojav, A., Demberei, S., et al. (2010). Denaturing gradient Gel elektroforeza analiza bakterii kwasu mlekowego i drożdży w tradycyjnym mongolskim mleku fermentowanym. Food Sci. Technol. Res. 16, 319-326. doi: 10.3136 / fstr.16.319
CrossRef Pełny tekst / Google Scholar
Mulder, N. J., and Apweiler, R. (2008). Baza danych InterPro i narzędzia do analizy domeny białka. Curr. Protokół. Bioinformatyka Rozdział 2, Unit2.7. doi: 10.1002 / 0471250953. bi0207s21
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Noguchi, H., Park, J., and Takagi, T. (2006). Metagen: prokariotyczne odnajdywanie genów z sekwencji genomu środowiskowego. Nucleic Acids Res. 34, 5623-5630. doi: 10.1093/nar / gkl723
PubMed Abstract / CrossRef Full Text / Google Scholar
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A. A., Korobeynikov, A., Lapidus, A. i in. (2013). Łączenie genomów jednokomórkowych i mini-metagenomów z chimerycznych produktów MDA. J. Comput. Biol. 20, 714–737. doi: 10.1089 / cmb.2013.0084
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Ott, A., Germond, J. E., and Chaintreau, A. (2000). Pochodzenie aldehydu octowego podczas fermentacji mleka przy użyciu prekursorów znakowanych (13) C. J. Agric. Chem.Żywności 48, 1512–1517. doi: 10.1021 / jf9904867
PubMed Abstract / CrossRef Full Text / Google Scholar
Pan, D. D., Zeng, X. Q., and Yan, Y. T. (2011). Charakterystyka Lactobacillus fermentum SM-7 wyizolowanego z koumiss, potencjalnej bakterii probiotycznej o działaniu obniżającym poziom cholesterolu. J. Sci. Jedzenie. Agric. 91, 512–518. doi: 10.1002 / jsfa.4214
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
Papadimitriou, K., Anastasiou, R., Maistrou, E., Plakas, T., Papandreou, N. C., Hamodrakas, S. J., et al. (2015). Pozyskanie poprzez poziomy transfer genów plazmidu pSMA198 przez Streptococcus macedonicus ACA-DC 198 wskazuje na mleczne pochodzenie gatunku. PLoS ONE 10: e0116337. doi: 10.1371 / dziennik.pone.0116337
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Segata, N., Waldron, L., Ballarini, A., Narasimhan, V., Jousson, O., and Huttenhower, C. (2012). Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Metody 9, 811-814. doi: 10.1038 / nmeth.2066
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Sun, Z., Liu, W., Zhang, J., Yu, J., Zhang, W., Cai, C., et al. (2010). Identyfikacja i charakterystyka dominujących bakterii lactobacilli wyizolowanych z koumiss w Chinach. J. Gen. Appl. Mikrobiol. 56, 257–265. doi: 10.2323 / jgam.56.257
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
Thompson, J. (1879). Wartość kumissa w wyniszczaniu chorób. Br. Med. J. 1, 270. doi: 10.1136 / bmj.1.1466.270-c
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Vincent, S. J., Faber, E. J., Neeser, J. R., Stingele, F., and Kamerling, J. P. (2001). Struktura i właściwości egzopolisacharydu wytwarzanego przez Streptococcus macedonicus Sc136. Glycobiology 11, 131-139. doi: 10.1093/glycob / 11.2.131
PubMed Abstract / CrossRef Full Text / Google Scholar
(2013)………………….. Zwiększona obfitość Sutterella spp. i przeżuwaczy w kale dzieci ze spektrum zaburzeń autyzmu. Mol. Autyzm 4, 42. doi: 10.1186/2040-2392-4-42
PubMed Abstract / CrossRef Pełny tekst / Google Scholar
Ward, T. L., Hosid, S., Ioshikhes, I., and Altosaar, I. (2013). Metagenom mleka ludzkiego: analiza zdolności funkcjonalnych. BMC Microbiol. 13:116. doi: 10.1186/1471-2180-13-116
PubMed Abstract / CrossRef Pełny tekst / Google Scholar
Watanabe, K., Fujimoto, J., Tomii, Y., Sasamoto, M., Makino, H., Kudo, Y., et al. (2009). Lactobacillus kisonensis Sp. listopad, Lactobacillus otakiensis Sp. listopad, Lactobacillus rapi Sp. z o. o. listopad oraz Lactobacillus sunkii Sp. listopad, heterofermentatywny gatunek wyizolowany z suńki, tradycyjnego japońskiego ogórka. Int. J. Syst. Evol. Mikrobiol. 59 (Pt 4), 754-760. doi: 10.1099 / ijs.0.004689-0
PubMed Abstract / CrossRef Full Text / Google Scholar
Wu, R., Wang, L., Wang, J., Li, H., Menghe, B., Wu, J., et al. (2009). Izolacja i wstępna selekcja probiotyczna bakterii lactobacilli z kumiss w Mongolii Wewnętrznej. J. Basic Microbiol. 49, 318–326. doi: 10.1002 / jobm.200800047
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Zerbino, D. R., and Birney, E. (2008). Velvet: algorytmy de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821-829. doi: 10.1101 / gr.074492.107
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Zhang, W., Sun, Z., Sun, T., and Zhang, H. (2010a). Badanie PCR i analiza sekwencji klastrów iol w szczepach Lactobacillus casei wyizolowanych z koumiss. Folia Microbiol. (Praha) 55, 603-606. doi: 10.1007 / s12223-010-0097-3
PubMed Abstract / CrossRef Pełny tekst / Google Scholar
Zhang, W., Yu, D., Sun, Z., Wu, R., Chen, X., Chen, W., et al. (2010b). Kompletna sekwencja genomu Lactobacillus casei Zhang, nowego probiotycznego szczepu wyizolowanego z tradycyjnych domowych koumiss w Mongolii Wewnętrznej w Chinach. J. Bakteriol. 192, 5268–5269. doi: 10.1128 / JB.00802-10
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Zhang, W., and Zhang, H. (2011). „Fermentacja i koumiss”, w Handbook of Food and Beverage Fermentation Technology, 2nd Edn, ed. Y. H. Hui (Boca Raton, FL: CRC Press), 165-172.
Google Scholar
Zhang, W. Y., Chen, Y. F., Zhao, W. J., Kwok, L. Y., and Zhang, H. P. (2015). Ekspresja genu układu proteolitycznego Lactobacillus helveticus H9 podczas fermentacji mleka. Ann. Mikrobiol. 65, 1171–1175. doi: 10.3168 / jds.2014-8520
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Zhao, W., Chen, Y., Sun, Z., Wang, J., Zhou, Z., Sun, T., et al. (2011). Pełna sekwencja genomu Lactobacillus helveticus H10. J. Bakteriol. 193, 2666–2667. doi: 10.1128 / JB.00166-11
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar